SILVER, Ag (At : 107.868) Silver is a white, malleable, and ductile metal. It has a high density (10.5 g ml-1) and melts at 96.5oC. It is insoluble in hydrochloric, dilute sulphuric (M) or dilute nitric (2M) acid. In more concentrated nitric acid (8M) (a) or in hot, concentrated sulphuric acid (b) it dissolves:
Silver forms monovalent ion in solution, which is colourless. Silver (II) compounds are unstable, but play an important role in silver-catalysed oxidation reduction processes. Silver nitrate is readily soluble in water, silver acetate, nitrite and sulphate are less soluble, while all the other silver compounds are practically insoluble. Silver complexes are however soluble, Silver halides are sensitive to light; these characteristics are widely utilized in photography.
Reactions of silver (I) ions: A solution of silver nitrate (0.1M) can be used to study these reactions.
1. Dilute hydrochloric acid: (or soluble chlorides): white precipitate of silver chloride
With concentrated hydrochloric acid precipitation does not occur. Decanting the liquid from over the precipitate, it dissolves in concentrated hydrochloric acid, when a dichloroargentate complex is formed:
By diluting with water, the equlibrium shifts back to the left and the precipitate reappears.
Dilute ammonia solution dissolves the precipitate, when diammineargentate complex ion is formed:
Dilute nitric acid or hydrochloric acid neutralizes the excess ammonia, and the precipitate reappears because the equilibrium is shifted back towards left.
Potassium cyanide(POISON) : dissolves the precipitate with formation of the dicyanoargentate complex:
The safest way to study this reaction is as follows: decant the liquid from the Precipitate, and wash it 2-3 time with water by decantation. Then apply e reagent.
Sodium thiosulphate dissolves the precipitate with the formation of dithiosulphatoargentate complex:
This reaction takes place when fixing photographic negatives or positive prints after development.
Sunlight or ultraviolet irradiation decomposes the silver chloride precipitate which turns to greyish or black owing to the formation of silver metal:
The reaction is slow and the actual reaction mechanism is very complicated. Other silver halides show similar behaviour. Photography is based on these reactions. In the camera these processes are only initiated the photographic material has to be Developed to complete the reaction.
Greyish or black silver particles appear on places irradiated by light; a negative image of the object is therefore obtained, The excess of silver halide has to be removed (to make the developed negative insensitive to light) by fixation.
2. Hydrogen sulphide: (gas or saturated aqueous solution) in neutral or acidic medium : back precipitate of silver sulphide
Hot concentrate nitric acid decomposes the silver sulphide, and sulphur remains in the form of white precipitate.
The reaction can be understood better if written in two steps:
If the mixture is heated with concentrated nitric acid for a considerable time sulphur is oxidized to sulphate and the precipitate disappears:
The precipitate is insoluble in ammonium sulphide, ammonium polysulphide, ammonia, potassium cyanide, or sodium thiosulphate. Silver sulphide can be precipitated from solutions containing diammine-, dicyanato- or dithiosulphato argentate complexes with hydrogen sulphide,
3. Ammonia solution: brown precipitate of silver oxide
The reaction reaches an equilibrium and therefore precipitation is incomplete at any stage. (If ammonium nitrate is present in the original solution or the solution is strongly acidic no precipitation occurs) The precipitate dissolves in excess of the reagent, and diammineargentate complex ions are formed:
The solution should be diposed of quickly, because when set aside silver nitride Ag3N precipitate is formed, which explodes readily even in a wet form.
4. Sodium hydroxide: brown precipitate of silver oxide:
A well-washed suspension of the precipitate shows a slight alkaline reaction owing to the hydrolysis equilibrium:
The precipitate is insoluble in excess reagent.
The precipitate dissolves in ammonia solution (a) and in nitric acid (b):
5. Potassium iodide: yellow precipitate of silver iodide
The precipitate is insoluble in dilute or concentrated ammonia, but dissolve readily in potassium cyanide (POISON) (a) and in sodium thiosulphate(b):
6. Potassium chromate in neutral solution: red precipitate of silver chromate
Spot test: place a drop of the test solution on a watch glass or on a spot plate, add a drop of ammonium carbonate solution and stir (this renders any mercury(I) or lead ions unreactive by precipitation as the highly insoluble carbonates). Remove one drop of the clear liquid and place it on drop-reaction Paper together with a drop of the potassium chromate reagent. A red ring of silver chromate is obtained.
Te reaction can be used for microscopic test, when a piece of potassium chromate crystal has to be dropped into the test solution. The formation of needle-like red crystals of silver chromate can be observed distinctly.
The precipitate is soluble in dilute nitric acid (a) and in ammonia solution (b):
The acidified solution turns to orange because of the formation of dichromate : ions in reaction (a).
7. Potassium cyanide(POISON) when added dropwise to a neutral solution of silver nitrate: white precipitate of silver cyanide:
When potassium cyanide is added in excess, the precipitate disappears owing to the formation of dicyanoargentate ions.
8. Sodium carbonate: yellowish-white precipitate of silver carbonate:
When heating, the precipitate decomposes and brown silver oxide precipitate is formed:
Nitric acid (a) and ammonia solution (b) dissolve the precipitate
Carbon dioxide gas is evolved in reaction (a).
9. Disodium hydrogen phosphate in neutral solution: yellow precipitate of silver Phosphate:
Nitric acid (a) and ammonia solution (b) dissolve the precipitate:
Phosphoric acid, formed in reaction (a) is a medium-strong acid, which is only slightly dissociated if nitric acid is present in excess.
10. Hydrazine sulphate (saturated): when added to a solution of diammineargentate ions, forms finely divided silver metal, while gaseous nitrogen is evolving:
If the vessel in which the reaction is carried out is clean, silver adheres to the glass walls forming an attractive mirror.
MERCURY, Hg (At: 200.59) – MERCURY(I) Mercury is a silver white, liquid metal at ordinary temperatures and has a density of 13.534gml-1 at 25oC. It is unaffected when treated with hydrochloric or dilute sulphuric : acid (2M), but reacts readily with nitric acid. Cold, medium concentrated (8m) nitric acid with an excess of mercury yields mercury(I) ions:
with an excess of hot concentrated nitric acid mercury(II) ions are formed:
Hot, concentrated sulphuric acid dissolves mercury as well. The product is mercury(I) ion if mercury is in excess
while if the acid is in excess, mercury(II) ions are formed:
The two ions, mercury(I) and mercury(II) behave quite differently against reagents used in qualitative analysis, and hence belong to two different analytical groups.
Reactions of mercury (I) ions: A solution of mercury(I) nitrate (0.05m) can be used for the study of these reactions.
1. Dilute hydrochloric acid or soluble chlorides: white precipitate of mercury(I) chloride (calomel) The precipitate is insoluble in dilute acids. Ammonia solution converts the precipitate into a mixture of mercury(II)amidochloride and mercury metal, both insoluble precipitates:
the reaction involves disproportionation, mercury(I) is converted partly to mercury(II) and partly to mercury metal. This reaction can be used to differentiate mercury(I) ions from lead(II) and silver(I). The mercury(II)amidochoride is a white precipitate, but the finely divided mercury makes it shiny black. The name calomel, coming from Greek (καονμελασ = nice black) refers to this characteristic of the originally white mercury(I) chloride precipitate. Mercury(I) chloride dissolves in aqua regia, forming undissociated but soluble mercury(II) chloride:
2. Hydrogen sulphide in neutral or dilute acid medium: black precipitate, which is a mixture of mercury (II) sulphide and mercury metal
Owing to the extremely low solubility product of mercury (Il) sulphide the reaction is very sensitive. Sodium sulphide (colourless). dissolves the mercury (II) sulphide (but leaves mercury metal) and a disulphomercurate (II) complex is formed:
After removing the mercury metal by filtration, black mercury (II) sulphide can again be precipitated by acidification with dilute mineral acids:
Sodium disulphide (yellow) dissolves both mercury (II) and mercury (II) sulphide:
This rather complicated reaction can be understood more easily by breaking it down into the following steps: First mercury is oxidized by the disulphide, yielding mercury (II)sulphide and (mono) sulphide ions:
Mercury (II) sulphide then dissolves in the (mono) sulphide formed in the previous reaction
Mercury (II) sulphide, which was originally present in the precipitate, reacts with disulphide ions yielding disulphomercurate(II) and trisulphide ions:
Combining the reactions (a), (b) and (c) together we obtain the reaction described above. Aqua regia dissolves the precipitate, yielding undissociatedmercury(II) chloride and sulphur:
This reaction can be understood as the sum of the following steps: When making up aqua regia chlorine atoms are formed:
These react partly with mercury, forming mercury(Il) chloride:
Another part of chlorine reacts with mercury(II) sulphide
Combination of 4(a)+ 3(b)+ 3(c) yields the equation
When heated with aqua regia, sulphur is oxidized to sulphuric acid and the solution becomes clear:
3. Ammonia solution: back precipitate which is a mixture of mercury metal and basic mercury(II) amidonitrate, (which itself is a white precipitate)
This reaction can be used to differentiate between mercury(I)and mercury(II) ions.
4. Sodium hydroxide: black precipitate of mercury(I) oxide
The precipitate is insoluble in excess reagent, but dissolves readily in dilute nitric acid. When boiling, the colour of the precipitate turns to grey, owing to disproportionation, when mercury(II) oxide and mercury metal are formed:
5. Potassium chromate in hot solution : red crystalline precipitate of mercury(I) chromate
If the test is carried out in cold, a brown amorphous precipitate is formed with an undefined composition. When heating the precipitate turns to red, crystalline mercury(I) chromate. Sodium hydroxide turns the precipitate into black mercury(I) oxide:
6. Potassium iodide, added slowly in cold solution: green precipitate of mercury (I) iodide
If excess reagent is added a disproportionation reaction takes place, soluble tetraiodomercurate(Il) ions and a black precipitate of finely divided mercury being formed:
When boiling the mercury(I) iodide precipitate with water, disproportionation again takes place, and a mixture of red mercury(II) iodide precipitate and finely distributed black mercury is formed:
7. Sodium carbonate in cold solution: yellow precipitate of mercury (I) carbonate:
The precipitate turns slowly to blackish grey, when mercury (II) oxide and mercury are formed:
The decomposition can be speeded up by heating the mixture.
8. Disodium hydrogen phosphate: white precipitate of mercury(I) hydrogen phosphate:
Mercury(II) cyanide, though soluble, is practically undissociated.
10. Tin(II) chloride: reduces mercury(I) ions to mercury metal, which appears in the form of a greyish-black precipitate:
Mercury(II) ions react in a similar way.
11. Potassium nitrite: reduces mercury metal from a solution of mercury(I) ions in cold, in the form of a greyish-black precipitate:
Under similar circumstances mercury(II) ions do not react. The spot test technique is as follows. Place a drop of the faintly acid test solution upon drop-reaction paper and add a drop of 50 per cent potassium nitrite solution, A black (or dark grey) spot is produced, The test is highly selective. Coloured ions yield a brown colouration which may be washed away, leaving the black spot.
12. Glossy copper sheet or copper coin: If a drop of mercury(I) nitrate is placed on a glossy copper surface, a deposit of mercury metal is formed:
Rinsing drying. And rubbing the surface with a dry cloth, a glittery, silverish spot is obtained. Heating the spot in a Bunsen-flame, mercury evaporates and the red copper surface become visible again, Mercury(II) solutions react in a similar way.
13. Aluminium sheet: If a drop of mercury(I) nitrate is placed on a clean aluminium surface, aluminium amalgam is formed and aluminium ions pass into solution:
The aluminiun which is dissolved in the amalgam is oxidized rapidly by the oxygen of the air and a voluminous precipitate of aluminiun hydroxide is formed. The remaining mercury amalgamates a further batch of aluminium, which again is oxidized thus considerable amounts of aluminium get corroded.
14. Dry test : All compounds of mercury when heated with a large excess (7-8 times the bulk) of anhydrous sodium carbonate in a small dry test-tube yield a grey mirror, consisting of fine drops of mercury, in the upper part of the tube. The globules coalesce when they are rubbed with a glass rod. Note : Mercury vapour is extremely poisonous, and not more than 0-1 gram of the substance should be used in the test.
Lead is poisonous element. If you have any idea about rat poison then you can link how poisonous Lead is. It is a bluish-grey metal with a high density (11.48 g ml-1 at room temperature). It readily dissolves in medium concentrated nitric acid (8M), and nitrogen oxide is formed also:
The colourles nitrogen oxide gas, when mixed with air, is oxidized to red- brown nitrogen dioxide:
2NO↑ (colourless) + O2 →+2NO2 ↑(red brown)
With concentrated nitric acid a protective film of lead nitrate is formed on the surface of the metal and prevents further dissolution. Dilute hydrochloric or sulphuric acid have little effect owing to the formation of insoluble lead chloride or sulphate on the surface.
Now Let’s see how to detect Lead by chemical reactions.
Reaction of lead (II) ions : A solution of lead nitrate (0.25M) or lead acetale (0.25) can be used for the study of these reactions.
1. Dilute hydrochloric acld (or soluble chlorides) : a white precipitate in cold and not too dilate solution:
The precipitate is soluble in hot water (33.4 g l-1 at 100o C while only 9.9 g l‑1 at 20o C), but separates again in long, needle-like crystals when cooling. It s also soluble in concentrated hydrochoric acid or concentrated potassium chloride when the tetrachloroplumbate(II) ion is formed:
If the precipitate is washed by decantation and dilute ammonia is added, no visble change occurs [difference from mercury (I) or silver ions], though a precipitate-exchange reaction takes place and lead hydroxide is formed:
2. Hydrogen sulphide in neutral or dilute acid medium: black precipitate of lead sulphide:
Precipitation is incomplete if strong mineral acids are present in more than 2M concentration. Because hydrogen ions are formed in the above reaction, it is advisable to buffer the mixture with sodium acetate.
Introducing hydrogen sulphide gas into a mixture which contains white lead chloride precipitate, the latter is converted into (black) lead sulphide in a precipitate-exchange reaction:
If the test is carried out in the presence of larger amounts of chloride [Potassium chloride (saturated), initially a red precipitate of lead sulpho- chloride is formed when introducing hydrogen sulphide gas:
This however decomposes on dilution (a) or on further addition of hydrogen sulphide (b) and black lead sulphide precipitate is formed:
Lead sulphide precipitate decomposes when concentrated nitric acid is added, and white, finely divided elementary sulphur is precipitated :
If the mixture is boiled, sulphur is oxidized by nitric acid to sulphate (a) which forms immediately white lead sulphate precipitate (b) with the lead ions in the solution.
Boiling lead sulphide with hydrogen peroxide (3%), the black precipitate turns white owing to the formation of lead sulphate:
The great insolubility of lead sulphide in water (4.9 x 10-11 g l-1) explains why hydrogen sulphide is such a sensitive reagent for the detection of lead, and why it can be detected in the filtrate from the separation of the sparingly soluble lead chloride in dilute hydrochloric acid.
3. Ammonia solution : white precipitate of lead hydroxide
The precipitate is insoluble in excess reagent.
4. Sodium hydroxide: white precipitate of lead hydroxide
The precipitate dissolves in excess reagent, when tetrahydroxoplumbate(1l) ions are formed :
Thus, lead hydroxide has an amphoteric character
Hydrogen peroxide (a) or ammonium peroxodisulphate (b), when added to a solution of tetrahydroxoplumbate (II) forms a black precipitate of lead dioxide by oxidizing bivalent lead to the tetravelent state :
5. Dilute suphuric acid (or soluble sulphates): white precipitate of lead sulphate :
The precipitate is insoluble in excess reagent. Hot, concentrated sulphuric acid dissolves the precipitate owing to formation of lead hydrogen sulphate:
Solubility is much lower in the presence of ethanol
Lead sulphate precipitate is soluble in more concentrated solutions of ammonium acetate (l0M) (a) or ammonium tartrate (6M) (b) in the presence of ammonia, when tetraacetatephumbate(I) and ditartratoplumbate(II) ions are formed:
The stabilities of these complexes are not very great; chromate ions, for example, can precipitate lead chromate from their solution.
When boiled with sodium carbonate the lead sulphate is transformed into lead carbonate in a precipitate-exchange reaction:
By washing the precipitate by decantation with hot water, sulphate ions can be removed and the precipitate will dissolve in dilute nitric acid
6. Potassium chromate in neutral acetic acid or ammonia solution : yellow precipitate of lead chromate
Nitric acid (a) or sodium hydroxide (b) dissolve the precipitate:
Both reactions are reversible: by buffering the solution with ammonia: acetic acid respectively, lead chromate precipitates again.
7. Potassium iodide : yellow precipitate of lead iodide
The precipitate is moderately soluble in boiling water to yield a colourless solution from which it separates on cooling in golden yellow plates.
An excess of a more concentrated (6M) solution of the reagent dissolves the precipitate and tetraiodoplumbat(II) ions are formed:
The reaction is reversible; on diluting with water the precipitate reappears.
8. Sodim sulphite in neutral solution: white precipitate of lead sulphite
The precipitate is less soluble than lead sulphate, though it can be dissolved by both dilute nitric acid (a) and sodium hydroxide (b).
9. Sodium carbonate: white precipitate of a mixture of lead carbonate and lead hydroxide
On boiling, no visible change takes place difference from mercury (I) and silver (I) ions. The precipitate dissolves in dilute nitric acid and even in acetic acid and CO2 gas is liberated:
10. Disodium hydrogen phosphate: white precipitate of lead phosphate
The reaction is reversible; strong acids (nitric acid) dissolve the precipitate.
The precipitate is also soluble in sodium hydroxide.
11. Potassium cyanide (POISON): white precipitate of lead cyanide
Which is insoluble in the excess of the reagent. This reaction can be used to distinguish lead (II) ions from mercury (I) and silver (I), which react in different ways.
Ea is more volatile than aluminium it will be discovered by spectral analysis
Atomic mass is 69.72
Melting point is 29. 75 degree centigrade.
Atomic volume is 11.8.
Oxidizes weakly upon heating to redness.
Decompose water at high temperature
Gives alums of the formula NH4 Ga(SO4)2 12 H2O
Ga is readily reduced by calcination of Ga2O3 in hydrogen inflow
Ga has been discovered by the spectroscopic method
Eka Aluminium properties were predicted by Mendeleev , while Ga properties are calculated using Modern Techniques .
Time of discovery of Gallium :
The time of discovery of gallium is known to an hour. “One Friday of August 27, 1875, between 3 p.m. and 4 p.m. I discovered some signs that there can be a new simple body in the by-product of chemical analysis of zinc blende from the Pierfitt mine in the Argele valley (Pyrenees).” With these words P. E. Lecoq de Boisbaudran began his report to the Paris Academy of Sciences.
He described some of the new element’s properties and noted that its presence in the ores was ascertained by spectra; analysis just as predicted by Mendeleev five years before. Boisbaudran extracted an extremely small amount of the substance and, therefore, could not study its properties properly.
On August 29, 1875 , Boisbaudran suggested to name the element “gallium” after Gaul, the ancient name of France. The scientist continued the investigation of the new element and obtained additional information which he included into his report to the Paris Academy and then sent it to be academic journal. In the middle of November the journal with the article reached Petersburg where Mendeleev was impatiently waiting for it. There is every reason to believe that Mendeleev had already learnt about gallium though at second hand. Two weeks earlier the Russian Chemical Society had received a report from Paris signed by P. de Clermont. It recounted the discovery of gallium and contained a brief description of its properties. However, it was much more important for Mendeleev to read what the discoverer himself had written. Mendeleev’s reaction was prompt; on November 16, he delivered a report to the Russian Physical Society. According to the minutes of the session, Mendeleev declared that the discovered metal was, most probably, eka-aluminium. Next day he wrote an article in French entitled “Note on the discovery of Gallium”. And finally, on November 18, Mendeleev spoke about gallium at a session of the Russian Chemical Society. Such a spurt of activity is understandable: the great chemist saw an element predicted by him becoming a reality. Mendeleev believed that if further investigation confirmed the similarity of eka-aluminium properties of those of gallium, this would be an instructive demonstration of the periodic law’s usefulness.
Six days later (a surprisingly short time!) the “Note on the Discovery of Gallium” appeared in the journal of the Paris Academy of Sciences. Boisbaudran’s reaction to it is of particular interest. He continued his experiments and prepared the new results for publication. The next article by the French scientist was published on December 6. As before, he complained of the difficulties caused by the extreme scarcity of gallium, described the preparation of the metal by the electrochemical method and discussed some of its properties, and suggested that the formula of gallium oxide had to be Ga2O3.
Only at the end of the article were there a few words about Mendeleev’s note. Boisbaudran admitted that he had read it with great interest since classification of simple substances interested him for a long time. He had never known about Mendeleev’s prediction of eka-aluminium properties but it did not matter; Boisbaudran believed that his discovery of gallium was facilitated by his own laws of spectral lines of elements with similar chemical properties. In his opinion, spectral analysis played a decisive role. And not a word that Mendeleev in his prediction of eka-aluminium also underlined the prominent role of spectral analysis in the discovery of the new element. According to Boisbaudran, Mendeleev’s predictions had nothing to do with the discovery of gallium.
However, as Boisbaudran went on studying the properties of metallic gallium and its compounds, his results continued to coincide with Mendeleev’s predictions. For instance, in May 1876, the Franch scientist established that gallium was readily fusible (its melting point is 29.5oC), its appearance remained the same after storage in air, and it was slightly oxidized when heated to redness. The same properties of eka-aluminium were predicted by Mendeleev in 1870, who calculated the density of eka-aluminium to be 5.9-6.0 on the basis of the periodic system and the densities of eka-aluminium’s neighbours. Lecoq de Boisbaudranm, however, making use of his spectral laws, found that the density of eka-aluminium was 4.7 and confirmed the value experimentally. Such a difference (less than two units) might seem small to a layman but it was essential for the future of the periodic law. Up to that time only qualitative characteristics of the predicted properties had been confirmed and density was the first quantitative parameter. And it turned out to be erroneous.
There is a widely known story that Mendeleev, having received Boisbaudran’s article citing a low (4.7) density of gallium, wrote him that the gallium obtained by the French chemist was contaminated most likely by sodium used in the process of gallium preparation. Sodium has a very low density (0.98), which could substantially decrease the density of gallium. Hence, it was required to purify gallium thoroughly.
This letter has not been found either in French or in the Mendeleev’s archives. There is only indirect evidence from Mendeleev’s daughter and the eminent historian of chemistry B. Menshutkin that the letter did exist. However, that may be Mendeleev’s views became known to Boisbaudran who decided to repeat the measurements of gallium’s density. This time he took into account that Mendeleev’s calculations for the hypothetical element’s density this time he took into account that Mendeleev’s calculations for the hypothetical element’s density gave 5.9. And be obtained this value at the beginning of September, 1876. His report about this fact needs no comments.
The French scientist became firmly convinced of the extreme importance of the confirmation of Mendeleev’s predictions about the density of the new element. Sometime later Lecoq de Boisbaudran send his photo to the great Russian chemist with the inscription: “With profound respect and an ardent wish to count Mendeleev among my friends. L. de B.”
Mendeleev wrote under it: “Lecoq de Boisbaudran. Paris. Discovered eka-aluminium in 1875 and named it “gallium”, Ga=69.7.”
In autumn 1879, F. Engels became acquainted with a new detailed chemistry textbook by H. Roscoe and C. Shorlemmer. For the first time it contained the story about the prediction of eka-aluminium by Mendeleev and its discovery as gallium. In an article to be later included in his Dialectics of Nature Engels quoted the corresponding text from the book and concluded: “by means of the unconscious application of Hegel’s law of the transformation of quantity into quality, Mendeleev achieved a scientific feat which is not too bold to put on a par with that of Leverrier in calculating the orbit of the still unknown planet Neptune”.
It is, perhaps, the only occasion in the history of transuranium elements that an element with a higher number (Z = 96) was identified earlier than its predecessor (Z = 95). In July 1944 the cyclotron of the University of California, which had already revealed to the world several synthesized elements, including plutonium, was geared to synthesize new transuranium elements. Seaborg and his coworkers bombarded a plutonium–239 target with accelerated alpha particles. One can readily reckon that as the alpha particle (the helium nucleus) has a charge of two the reaction product could be an isotope of element 96, provided that neutrons were emitted from the resulting nuclei. If the process mechanism was such that protons were emitted, rather than neutrons, then an isotope of element 95 could be synthesized. Indeed, various radioactive substances were produced in the plutonium target and it was difficult at first to identify “who was who”. Only skillful chemical analysis revealed that the mixture definitely contained the isotope 24296. To verify the discovery, the same isotope, plutonium–239, was bombarded with a high–intensity neutron beam so that the following chain of reactions took place :
After absorption of neutrons plutonium converted into element 95 via beta decay and this element absorbed a neutron and converted into element 96.
This final product was similar to that which the scientists had assumed to be the isotope of element 96 with a mass number of 242. The newly discovered element was named curium after the Curies. Another factor prompted this name. In the Mendeleev table element 96 was regarded as an analogue of gadolinium belonging to the rare–earth series the history of which had been started by J. Gadolin; in their turn, the Curies were the pioneers of the study of radioactivity whose development produced such amazing results. In January 1945 element 95 was extracted from plutonium bombarded with neutrons. The element was named americium in honour of America (and owing to its similarity to europium from the rare–earth series).
Though the researchers had accumulated considerable experience in syntheses the difficulties involved in producing americium and curium proved unusually great. It took a long time to distinguish definitely between americium–241 and curium–242. Both isotopes proved to be not the longest–lived ones. The longest–lived isotopes were americium–243 (a half–life of 7 950 years) and curium–247 (a half–life of 1.64 × 107 years), which were only synthesized in the fifties. The total of 11 americium isotopes and 13 curium isotopes are currently known. Here are a few more events in the history of these elements. Pure americium was extracted in 1945 and in 1951 it was prepared in a metallic form. The same year metallic curium was prepared.
The discovery of curium ends the first breakthrough period in the history of transuranium elements. The discoveries of neptunium, plutonium, americium, and curium were of great significance for science. It was for the first time that scientists artificially extended the boundaries of the periodic system. The properties of these elements proved to be quite different from those expected and chemists had to start seriously thinking how best to fit them into the periodic system.
Was it just a chance that polonium and radium were the first to be discovered among radioactive elements? The answer is apparently no. Owing to its long half–life radium can be accumulated in uranium ores. Polonium has a short half–life (138 days) but it emits characteristic high–intensity alpha radiation. Though the discovery of polonium gave rise to a controversy it soon died off.
The third success of the young science of radioactivity was the discovery of actinium. Soon after they had discovered radium the Curies suggested that uranium ore could contain other, still unknown radioactive elements. They entrusted their collaborator A. Debierne with verification of this idea.
Debierne started his work with a few hundred kilograms of uranium ore extracting the “active principle” from it. After he had extracted uranium, radium, and polonium he was left with a small amount of a substance whose activity was much higher than the activity of uranium (approximately, by a factor of 100 000). At first, Debierne assumed that this highly radioactive substance was similar to titanium in its chemical properties. Then he corrected himself and suggested a similarity with thorium. Later, in spring of 1899 he announced the discovery of a new element and called it actinium (from the Greek for radiation).
Any textbook, reference book or encyclopedia gives 1899 as the date of the discovery of actinium. But in fact, to say that in 1899 Debierne discovered a new radioactive element–actinium–means to ignore very significant evidence to the contrary.
The real actinium has little in common with thorium but we did not mean this chemical difference as evidence against the discovery of actinium by Debierne. The main argument is as follows. Debierne believed that actinium was alpha–active and its activity was 100 000 times that of uranium. Now we know that actinium is a mild beta–emitter, that is, it emits beta rays of a fairly low energy which are hot that easy to detect. Of course, the primitive radiometric apparatus of Debierne was not capable of doing it.
Then what did Debierne discover? It was a complex mixture of radioactive substance including actinium. But the weak beta radioactive of actinium was quite indistinguishable against the background of the alpha rays emitted by the products of actinium decay. It took several years to extract the real actinium from this mixture of radioactive products.
In 1911 the outstanding British radiochemist F. Soddy published a book entitled chemistry of Radioactive Elements where he described actinium as an almost unknown element. He wrote that its atomic weight was unknown, the mean life time was also unknown, it did not emit rays (this shows how difficult it was to detect the beta radiation of actinium), and its parent substance was unknown. In a word, much about actinium was still vague.
The evidence presented by Debierne for his discovery of actinium did not seem convincing to his contemporaries. It is no wonder that soon another scientist–the German chemist F. Giesel–claimed a discovery of a new radioactive element. He also extracted a certain radioactive substance whose properties were similar to those of the rare–earth element. This fact is closer to the truth in the light of our current knowledge. Giesel named the new element emanium because it evolved a radioactive gas–emanation–which made a zinc sulphide screen to glow. Along with the radiotellurium vs. polonium controversy there appeared a similar controversy between the supporters of actinium and emanium. The first controversy ended by establishing identity between the elements in question. The second controversy proved to be more complicated and could not be speedily resolved since the behaviour of the third new radioactive element was too wayward. The name of Debierne went into the historical records as the name of the discoverer of actinium. However, the substance extracted by Giesel contained a significant proportion of pure actinium as was shown later. Giesel also succeeded in observing the spectrum of emanium. Many scientists believed that they proved identity of actinium and emanium. Gradually, the controversy lost its edge.
The British radiochemist A. Cameron was the first (1909) to place the symbol Ac into the third group of the periodic system (actually, he was the first to put forward the name radiochemistry for the relevant science). But only in 1913 was the position of actinium in the periodic system established reliably. As increasingly pure actinium preparations were obtained the scientist encountered an amazing situation–the radiation emitted by actinium proved to be so weak that some scientists even doubted if it emits at all. It has even been suggested that actinium undergoes an entirely new, radiation less, transformation. It was only in 1935 that beta rays emitted by actinium were reliably detected. The half–life of actinium was found to be 21.6 years.
For a long time extraction of metallic actinium was just out of question. Indeed, one ton of pitchblende contains only 0.15 mg of actinium while the content of radium is as high as 400 mg. A few milligrams of metallic actinium were obtained only in 1953 after reduction of AcCl3 with potassium vapour.
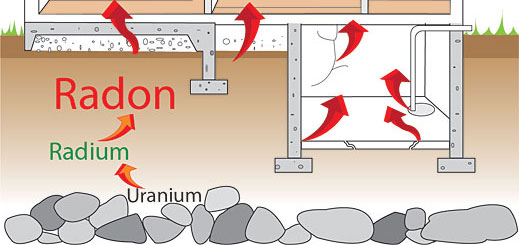
Radon Rn is the 86th element of the periodic system. It is the heaviest of the noble gases. It is highly radioactive and its natural abundance is so low that it could not be identified when W. Ramsay and M. Travers discovered other inert elements. Only application of the radiometric method made possible the discovery of radon.
What we know as radon at present is the combined name for the three natural isotopes of the element No. 86, which were discovered one by one and called emanations. Their appearance heralded a new stage in the studies of radioactivity as they were the first gaseous radioactive substances.
At the beginning of 1899 E. Rutherford (who lived at the time in Canada) and his collaborator R. Owens studied the activity of thorium compounds. Once Owens accidentally threw open the door to the laboratory where a routine experimenter was performed. There was a drought and the experimenters noticed that the intensity of radiation of the thorium preparations suddenly dropped. At first they ignored this event but later they observed that a slight movement of air seemed to remove a larger part of the activity of thorium. Rutherford and Owens decided that thorium continuously emitted a gaseous radioactive substance, which they called the emanation (from the Latin to flow) of thorium, or Theron.
By way of analogy, it was suggested that other radioactive elements could also evolve emanations. In 1900 the German physicist E. Dorn discovered the emanation of radium and three years later Debierne observed the emanation of actinium. Thus, two new radioactive elements were found, namely, radon and action. An important observation was that all the three emanations differed only in their half–lives–51.5 s for thoron, 3.8 days for radon, and 3.02 s for action. The longest–lived element is radon and therefore it was used in all studies of the nature of emanations. All the other properties of emanations were identical. All of them lacked chemical manifestations, that is, they were inert gases (analogues of argon and other noble gases). Later they were found to have different atomic masses. But there was just a single slot for these three elements in the periodic system, immediately below xenon.
Such exclusive situation soon became a rule. Therefore, we shall have to discuss briefly some important events in the history of radioactivity studies. Now we must finish the story of radon. This name remained because radon is the longest–lived element among the radioactive inert gases. Ramsay suggested to name it niton (from the Latin for glowing) but this name did not take root.
When the Curies and G. Bemont analysed pitchblende they noticed a higher radioactivity of one more fraction, apart from the bismuth fraction. After they had succeeded in extracting polonium they started to analyse the second fraction thinking that they could find yet another unknown radioactive element.
The new element was named radium from the Latin radius meaning ray. The birthday of radium was December 26, 1898. When the members of the Paris Academy of Sciences heard a report entitled “On a new highly radioactive substance contained in pitchblende”. The authors reported that they had managed to extract from the uranium ore tailings a substance containing a new element whose properties are very similar to those of barium. The amount of radium contained in barium chloride proved to be sufficient for recording its spectrum. This was done by the well–known French spectral analyst E. Demarcay who found a new line in the spectrum of the extracted substance. Thus, two methods–radiometry and spectroscopy–almost simultaneously substantiated the existence of a new radioactive element.
The position of radium among the natural radioactive elements (of course, excluding thorium uranium) almost immediately proved to be the most favourable one owing to many reasons. The half–life of radium was soon found to be fairly long, namely, 1 600 years. The content of radium in the uranium ores was much higher than that of polonium (4 300 times higher); this contributed to natural accumulation of radium. Furthermore, the intensity of alpha radiation of radium was sufficiently high to allow an easy monitoring of its behaviour in various chemical procedures. Finally, a distinguishing feature of radium was that it evolved a radioactive gas known as emanation (see p. 183). Radium was a convenient subject for studies owing to a favourable combination of its properties and therefore it became the first radioactive element (again, with the exception of uranium and thorium) to find its permanent place in the periodic system without long delay. Firstly, chemical and spectral studies of radium demonstrated that in all respects it belongs to the subgroup of alkaline earth metals; secondly, its relative atomic mass could be determined accurately enough. To do be obtained. The Curies worked ceaselessly for 45 months in their ill–equipped laboratory processing uranium ore tailings from Bohemian mines. They performed fractional crystallization about 10 000 times and finally obtained a priceless prize–0.1 g of radium chloride. The history of science knows no more noble examples of enthusiastic work. This amount was sufficient for measurements and on March 28, 1902, Marie Curie reported that the relative atomic mass of radium was 225.9 (which does not differ much from the current figure of 226.02). This value just suited the suggested position of radium in the periodic system.
The discovery of radium was the best substantiated one among the many alleged discoveries of radioactive elements, which soon followed. Every year more new discoveries were reported. Radium was also the first radioactive element obtained in the metallic form.
Marie Curie and her collaborator A. Debierne electrolyzed a solution containing 0.106 g of radium chloride. Metallic radium deposited on the mercury cathode forming amalgam. The amalgam was put into an iron vessel and heated under a hydrogen flow to remove mercury. Then grains of silvery whitish metal glistened at the bottom of the vessel.
The discovery of radium was one of the major triumphs of science. The studies of radium contributed to fundamental changes in our knowledge of the properties and structure of matter and gave rise to the concept of atomic energy. Finally, radium was also the first radioactive element to be practically used (for instance, in medicine).
Polonium was the first natural radioactive element discovered with the radiometric technique. Back in 1870 the main properties of polonium were predicted by D. I. Mendeleev. He wrote: “Among heavy metals we can expect to find an element similar to tellurium whose atomic weigh is greater than that of bismuth. It should possess metallic properties, and give rise to an acid whose composition and properties should be similar to those of sulphuric acid and whose oxidizing power is higher than that of telluric acid…
The oxide RO2 cannot be expected to have acidic properties which tellurous acid still has. This element will form organometallic compounds but not hydrogen compounds…”
Nineteen years had passed and Mendeleev made a significant addition to his description of dvi–tellurium (as he called the unknown element). He predicted the following properties: relative atomic mass 212; forms oxide DtO3; in a free state the element is a crystalline low–melting non–volatile metal of grey colour with a density of 9.8; the metal is easily oxidized to DtO2; the oxide will have weak acidic and basic properties: a hydride of the element, if it exists at all, must be unstable; the element must form alloys with other metals.
Below readers will see for themselves how accurate were Mendeleev’s predictions of the properties of a heavy analogue of tellurium. But these predictions had only an indirect effect on the history of polonium, if any. The discovery of polonium (and then radium) proved to be a significant milestone in the science of radioactivity and gave an impetus to its development.
As one can see from the laboratory log–book of Marie and Pierre Curie they started to study the Becquerel rays, or uranium rays, on December 16, 1897. First the work was conducted by Marie alone and then Pierre joined her on February 5, 1898. He performed measurements and processed the results. They mainly measured the radiation intensities of various uranium minerals and salts as well as metallic uranium. The results of extensive experiments suggested that uranium compounds had the lowest radioactivity, the metallic uranium exhibited a higher radioactivity, and the uranium ore known as pitchblende had the highest radioactivity. These results indicated that pitchblende, probably, contained an element whose activity was much higher than that of uranium.
As early as April 12, 1898 the Curies reported this hypothesis in the proceedings of the Paris Academy of Science. On April 14 the Curies started their search for the unknown element with the assistance of the chemist G. Bemont. By the middle of July they finished the analysis of pitchblende. They carefully measured the activity of each product successively isolated from the ore. Their attention was focussed on the fraction containing bismuth salts. The intensity of the rays emitted by this fraction was 400 times that of metallic uranium. If the unknown element really did exist it had to be present in this fraction.
Finally, on July 18 Marie and Pierre Curie delivered a report to a session of the Paris Academy of Science entitled “On a new radioactive substance contained in pitchblende”. They reported that they had managed to extract from pitchblende a very active Sulphur compound of a metal that had previously been unknown. According to its analytical properties it was a neighbour of bismuth. The Curies suggested, if the discovery could be proved, to name the new element in honour of the country where Marie had been born and brought up, that is, polonium after Poland.
The scientists emphasized that the element had been discovered with a new research method (the term “radioactivity”, which later became conventional, was first introduced in this report).
The introduction of spectral analysis made it possible to reveal the existence in natural objects of elements that could not be seen, felt or weighed. Now the history repeated itself but the role of indicator was played by radioactive radiation, which could be measured with a radiometric technique. However, the results of the Curies were not faultless. They were wrong in suggesting a chemical similarity between polonium and bismuth. Even a brief look at the periodic system shows that the existence of a heavy analogue of bismuth is hardly possible. But one must not forget that the Curies did not extract pure metal, could not determine its relative atomic mass, and, finally, did not see differences in the spectra of polonium and bismuth. This is why they actually ignored a possible analogy between polonium and tellurium.
Thus, we may regard 18 July, 1898, as the date of just a preliminary discovery of polonium as substantiation of the discovery took quite a long time. The high intensity of radiation from polonium made difficult its study. The radiation was found to consist of only alpha rays with no beta or gamma rays. A strange finding was that the activity of polonium decreased with time and the decrease was rather noticeable; neither thorium nor uranium exhibited such behaviour. This is why some scientists doubted whether polonium existed at all. The sceptics said it was just normal bismuth with traces of radioactive substances.
But in 1902 the German chemist W. Marckwald extracted the bismuth fraction from two tons of uranium ore. He put a bismuth rod into a bismuth chloride solution and observed precipitation of a highly radioactive substance on it which he took for a new element and named radiotellurium. Later he recalled: “I named this substance radiotellurium just for the time being since all its chemical properties suggested placing it into the sixth group into the still unoccupied box for the element with a somewhat higher atomic weight than that of bismuth…. The element was more electronegative than bismuth but more electropositive than tellurium; its oxide should also have basic rather than acidic properties.
All this corresponded to radiotellurium…. The expected atomic weight for this substance was about 210”. Later he said that he had got his idea for extracting polonium when analysing the periodic system.
As for the polonium discovered earlier Marckwald promptly declared it a mixture of several radioactive elements. This led to a stormy discussion of the real nature of polonium and radiotellurium. Most scientists supported the Curies. A. later comparison of the two elements revealed their identity. The discovery was credited to the Curies and the name “polonium” was retained.
Though polonium was the first of the new natural radioactive elements its symbol Po did not appear in the appropriate box in the periodic system. The atomic mass of the element was very difficult to measure. The lines of the polonium spectrum were reliably identified in 1910. It was only in 1912 that the symbol Po occupied its place in the periodic table.
For almost half a century scientists had to be satisfied to work only with polonium compounds (usually in rather small amounts). The pure metal was prepared only in 1946. High density layers of metallic polonium prepared by vacuum sublimation have a silvery colour. Polonium is a pliable low–melting metal (melting point 254oC, boiling point 962oC), its density is about 9.3 g/cm3. When polonium is heated in the air it readily forms a stable oxide; its basic and acidic properties are weakly manifested. Polonium hydride is unstable. Polonium forms organometallic compounds and alloys with many metals (Pb, Hg, Ca, Zn, Na, Pt, Ag, Ni, Be). When we compare Mendeleev’s predictions with these properties we see how close they are to the truth.
As regards history, rhenium had an undoubted advantage over hafnium: nobody had ever questioned the fact that element No. 75 had to be an analogue of manganese, or tri-manganese in Mendeleev’s terminology. However, in all other respects there was no certainty.
Let us perform an experiment. If we select at random a few monographs and textbooks where rhenium is discussed we shall see that the authors agree on some things while sharply disagreeing on others. They all agree that rhenium was discovered in 1925 but when it comes to the source from which rhenium was extracted, they disagree. Among minerals mentioned as sources of rhenium are columbite and platinum ore, native platinum and tantalite, niobite and wolframite, alvite and gadolinite. Even an experienced geochemist will be at a difficulty finding his way among so varied a group of minerals.
After these introductory remarks, we may name the discoverers of rhenium: V. Noddack, I. Takke (who later married V. Noddack), and the spectroscopist O. Berg. Their authorship was never contested by anybody. This may be the only case when engineers became interested in the yet undiscovered element. They were aware of the uses of the periodic system. Since tungsten was widely used in electrical engineering, there was every reason to believe that element No. 75 would possess properties even more valuable for this industry. It is highly probable that the first attempts of the Noddacks to find this element were prompted by practical needs.
In 1922, after thorough preparations they set to work. First of all, they collected all reports on the discovery of manganese analogues. Since these discoveries remained unconfirmed, it was tempting to check them. The scientists drew up an extensive program of research: they were going to look for two elements at once since unknown manganese analogues included not only element No. 75 but also its lighter predecessor–element No. 43 with an unusual fate (see p. 200). The periodic table made it possible to predict many of their properties. We can now compare the Noddacks’ predictions on rhenium with the actual properties of the element.
Prediction Modern data
Atomic mass 187-188 186.2
Density 21 20.5
Melting point 3300 K 3 323 K
The higher oxide formula X2O7 Re2O7
Melting point of the higher
Oxide 400-500oC 220oC
The agreement is, indeed, excellent. Only the melting pint of the oxide proved to be much lower that
the expected one whereas on the whole Mendeleev’s classical method of prediction was fully confirmed. In other words the Noddacks had a perfectly good idea about what element No. 75 (and element No. 43) was going to be. Thus, the history of rhenium was closely related to the history of its light analogue.
But where to search for these element? Predicting the geochemical behavior of rhenium the Noddacks used to the full the capacity of theoretical geochemistry of that time; They even knew that it had to be a very rare element. They could not know, however, that it was a trace element and that, therefore, what seemed unquestionable to them was in effect open to doubt.
The scientists planned to investigate two groups of minerals: platinum ores and so called columbites (tantalites). Four years (from 1921–1925) were spent in searching for the wanted elements but in vain. Then a communication appeared about the discovery of hafnium whose existence in nature was proved by X-ray spectroscopy. Undoubtedly, this event gave the Noddacks the idea to use the same method in order to prove the existence of manganese analogues and they turned for help to O. Berg, a specialist in X-ray spectroscopy.
In June 1925, V. Noddacks, I. Takke, and O. Berg published an article about the discovery of two missing elements, Masurium (No. 43) and rhenium (No. 75). They were found in columbite and in the Uralian platinum and named after two German provinces. The elements X-ray spectra provided the main confirmation of their existence; but there was no question of extracting the elements and the reasoning of the German scientists was, in general, too involved. However, the article attracted attention and other scientists tried to reproduce the results.
However, no such reproduction followed. A year passed and the Soviet scientist O. E. Zvyagintsev and his colleagues proved irrefutably that the Uralian platinum ore contained no new elements. After that the German scientists continued to study columbites which varied considerably in composition but, according to the predictions, had to contain mysterious manganese analogues. They subjective the minerals to complex chemical treatment in order to concentrate the unknown elements and performed X-ray spectral analysis. The data obtained were reassuring but definite conclusions would have been premature: the scientists could not obtain any noticeable amounts of elements No. 43 and No. 75 and experimentally determine their properties.
Nobody could reproduce the results obtained by the Noddacks. Their compatriot W. Prandtl even sent his assistant. A Grimm to the Noddacks’ laboratory to watch them prepare manganese analogues Back home, A. Grimm reproduced the entire procedure, perfected it and…, we do not know the extent of his distress about the wasted time. The English scientists F. Loring and the Czechs Ya. Geirovskii and Y. Druce also doubted the Noddacks’ results. Later, Loring, Geirovskii, and druce claimed the priority of discovering element No. 75 by other methods and from other sources. History has retained their names but not as discoverers of rhenium.
The two German scientists believed to have also isolated element No. 43 (known later as technetium). Now we know that they by no means could detect the presence of technetium at the time but, nevertheless, the Noddacks were more sure of its discovery than of the discovery of rhenium (the fact which is hardly a feather in their cap). As time passed, the Noddacks became convinced that the range of the minerals for analysis had to be considerably enlarged. The previous geochemical prediction did not, apparently, come true. In the summer of 1926 and in 1927 the Noddacks went to Norway to collect minerals among which were: tantalite, gadolinite, alvite, fergusonite, and molybdenite. In the early 1928 the scientists, analysing the minerals, isolated about 120 mg of rhenium mainly from molybdenite (molybdenum sulphide). Earlier it had never been considered as a possible source of manganese analogues.
Thus, rhenium became, at last, a reality. An end was put to doubts and the symbol Re occupied forever box No. 75 in the periodic table; masurium, however, remained an enigma for a long time.
Hence, 1928 is the date of the reliable discovery of rhenium, the final step in the long process of search. As regards the widely accepted date, 1925, it is only a landmark in the prehistory of the element.
Having planned the directions of research, the Noddacks assembled all publications of supposed discoveries of eka-manganese. Their notes were lost during the second World War but, undoubtedly, the name of the Russian Scientists S. F Kern and the name of the element “devium” were mentioned in them. This may be the most reliable discovery of a new element of all unreliable discoveries. And it is equally possible that the history of element No.75 could have begun 50 years earlier.
The events were as follows. In 1877 reports appeared about the discovery of a new metal “devium” named after H. Davy. The reports aroused great interest and Mendeleev suggested inviting S. F.Kern to report to a session of the Russian Chemical Society. The scientists of Bunsen’s laboratory in Heidelberg decided to check Kern’s result carefully. Later his results were confirmed by two or three other scientists the most interesting fact was that some chemical reactions proved to be identical to those found later for rhenium. Does not it point to the identity of devium and rhenium?
For some reason or other S. F Kern lost interest in his discovery and never returned to the problem after 1878. He had extracted the element from platinum ores, which seems impossible from modern point of view (recall Zvyagintsev’s work in 1926). The fact is, however, that platinum ores have a complex and varied composition. The Uralian ore does not contain rhenium but its presence as traces in ores of other deposits has been proven.
F. Kern studied a very rare sample of platinum ore from Borneo where by that time mines had already been abandoned. At the beginning of the 20th century the Russian chemist G. Chernik worked on the island. Analyzing platinum ores he found a constant mass loss in all samples and tried to explain it by the presence of an unknown element. This element- could well be Kern’s “devium”.
In 1950 Y. Druce devoted a large article to devium. He wrote that if rhenium would be discovered in platinum minerals, this would confirm Kern’s discovery. Samples of platinum ores from Borneo can be found now only in a few mineralogical museums of the world. It would be of interest to analyse them thoroughly. This is a case when the history of a chemical element could be partially changed.
The Institute of Theoretical Physics of the Copenhagen University in Denmark was the birthplace of a new element with Z = 72; the date of birth was the end of December, 1992, although the article about the discovery appeared in a scientific journal only in January, 1923. The Dutch spectroscopist D. Coster and the Hungarian radiochemist G. Hevesy named the element after the ancient name of CopenhagenHafnia. N. Bohr, whose role in the discovery of hafnium was decisive, stood at the cradle of the element.
The source of element No. 72 was zircon, a rather common mineral, consisting mainly of zirconium oxide. And it was Bohr who suggested the mineral as a subject of investigation. Why was the Dutch physicist so confident of success? Let us go back to the 1870’s when Mendeleev was drawing up his periodic system. He reserved the box under zirconium for an unknown element with the atomic mass about 180. Using Mendeleev’s terminology, we could name it eka-zirconium. After Mendeleev’s predictions of gallium, scandium, and germanium had come true, the confidence in the existence of eka-zirconium became stronger. The question, however, remained about the properties of this hypothetical element. Mendeleev refrained from definite assessments. Generally speaking, there were two possibilities: either eka-zirconium was part of the IV B-subgroup of the periodic table, i.e. an analogue of zirconium, or it belonged to the rare-earth family as its heaviest element. Now the time has come to recall the name “celtium” (see p. 138).
Having split ytterbium and separated lutetium, the last of the REEs existing in nature, G. Urbain continued the difficult work of separating heavy rare earths. Finally, he succeeded in collecting the fraction whose optical spectrum contained new lines. This event took place in 1911 but at the time did not attract the attention of the scientific community. Perhaps Urbain himself, having suggested the name for it, was not quite sure that he had really discovered a new element. At any rate, he thought it wise to send samples of celtium to Oxford where Moseley worked. Moseley studied the samples by X-ray spectroscopy but the X-ray photographs turned out to be of a poor quality. Nevertheless, in August 1914, Moseley published a communication in which he firmly stated that celium was a mixture of known rare earths. The communication remained practically unnoticed. In a word, the discovery of celtium for a very long time considered to be doubtful, although the symbol Ct sometimes appeared in scientific journals.
Meanwhile N. Bohr was working on the theory of electron shells in atoms which also became the corner-stone of the periodic system theory and, at last, explained the periodic changes in the properties of chemical elements. Bohr also solved the problem which had interested chemists of many years: he found the exact number of rare-earth elements. There had to be fifteen of them from lanthanum to lutetium. Only one REE between neodymium and samarium (later known as promethium, see p.208) remained unknown. Bohr came to this conclusion on the basis of the laws found by him which governed the formation of electron shells of atoms with increasing Z.
Thus, if celtium were indeed a rare-earth element, Bohr’s theory would eliminate it completely. Why couldn’t it be eka-zirconium? Having proved that lutetium completed the REE series, Bohr firmly established that element No. 72 had to be a zirconium analogue and could be nothing else. Bohr advised D. Coster and G. Hevesy to look for the missing element in zirconium minerals. Now all this seems to us quite logical and clear but at that time many things were at stake: if element No. 72 could not be proved to be a complete analogue of zirconium, the whole of Bohr’s periodic system theory would have been questioned. Having separated hafnium from zirconium Coster and Hevesy confirmed this theory experimentally just as the discovery of gallium had been a confirmation of Mendeleev’s periodic system than half a century before.
When Urbain read the communication about the discovery of hafnium, he understood that this was the end of celtium. Not everybody can take the bitterness of defect with dignity. Urbain was reluctant to part with celtium and continued his attempts to identify it with element No.72. The French spectroscopist A. Dauvillier came to help; he tried to prove the originality of celtium spectra thus making the “element” one of the rare earths.
Moreover, Urbain and Dauvillier declared that Coster and Hevesy had only rediscovered celtium but nothing much came of it, since hafnium soon came into its own. It was prepared in pure form and new spectral investigations showed that there was nothing in common between hafnium and celtium. What an irony of history! Urbain had everything to be the first to discover hafnium. At the beginning of 1992 he and his colleague C. Boulange analysed thortveitite, a very rare mineral from Madagascar. The mineral contained 8 per cent of zirconium oxide and the content of hafnium oxide was even higher. It is the only case when hafnium is contained in the mineral in amounts greater than those of zirconium and, nevertheless, Urbain and Boulange failed to uncover element No. 72. The reason for this lies in the great chemical similarity between zirconium and hafnium.
The history of gallium, scandium and germanium shows that their discoveries were practically unaffected by the periodic law and periodic system. However, the properties predicted by D.I Mendeleev for eka-aluminium, eka-boron and eka-silicon coincided with those of gallium, scandium, and germanium. Mendeleev had determined the main features of these elements long before they were discovered in nature. Is not this fact a striking evidence of the periodic system’s power of prediction?
The discovery of gallium and its identity with eka-aluminium became milestones in the history of the periodic law and in the history of discovery of elements. After 1875 even those scientists who had disregarded the periodic system had to recognize its value. And among them there were top researchers, such as R. Bunsen, the creator of spectral analysis (he once said that to classify elements is the same thing as to search for regularities in the stock-exchange quotations) or P. Cleve who had never mentioned the periodic system in his lectures. The discovery of scandium and germanium meant further triumph of Mendeleev’s theory of periodicity.
In addition to the classic triad Mendeleev predicted the existence other unknown elements. On the whole, as early as 1870 Mendeleev saw about ten vacant places in his table. He saw them, for instance, in the seventh group where there were neither manganese analogues nor a heavy iodine’s analogue (the heaviest halogen which had to possess metallic properties).
In Mendeleev’s papers we find mention of eka-, dvu-, and tri-manganese and eka-iodine. The scientist firmly believed in their existence. And here we encounter a very interesting fact in the predictions. Eka-manganese (known subsequently as technetium) and eka–iodine (astatine) were synthesized later. Mendeleev, naturally, could not know that they did not exist in nature and firmly believed in their existence since these elements filled in the gaps in the periodic system and made it more logical.
The prediction consists of two stages: prediction of the existence of an element and prediction of its main properties. The first stage was in many respects guess-work for Mendeleev. As yet unknown was the phenomenon of radioactivity making some elements so short-lived that their earthly existence is impossible at all or they exist only because they are products of radioactive transformations of long-lived elements (thorium and uranium).
The second stage was completely within Mendeleev’s power and depended on his confidence. Sometimes Mendeleev predicted boldly and resolutely. This was the case with eka-aluminum, eka-boron, and eka-silicon: these elements had to be placed in that part of the periodic table where many well-known and well-studied elements had already been located–the region of reliable prediction. Sometimes Mendeleev predicted the properties of unknown elements with the extreme caution. Among them were analogues of manganese, iodine, and tellurium as well as the missing elements of the beginning of the seventh period: eka-cesium, eka-barium, eka-lanthanum, and eka-tantalum. Here Mendeleev was groping in the dark, darling only to estimate atomic masses and suggest formulas of oxides. Mendeleev thought that it was difficult to predict the properties of the unknown elements (including those of REEs) whose places were at the boundaries of the system because there were few known elements around them. This was the “grey” area of uncertain prediction. Of course, they included the rare–earth elements. Finally, in some parts of the periodic table prediction was completely unreliable. They included those mysterious stretches extending in the directions of hypothetical elements lighter than hydrogen and heavier than uranium. Mendeleev never thought that the periodic system had to begin with hydrogen. He even wrote a paper in which he described two elements preceding hydrogen. Only when physicists explained the meaning of the periodic law, his mistake became clear: the nucleus of the hydrogen atom had the smallest charge equal to 1. As regards elements which are heavier than uranium, Mendeleev conceded the existence of a very restricted number of them and never took the liberty of predicting, even approximately, their possible properties. Predictions of this kind did not come until much later when they signaled important events in the history of science.
Among the three elements predicted by Mendeleev eks-silicon was the last to be discovered and its discovery was to a greater extent than in the case of the two others, due to a chance. Indeed, the discovery of gallium by P Lecoq de Boisbaudran was directly related to his spectroscopic investigations, and the separation of scandium by L. Nilson and P. Cleve was associated with thorough investigation of REEs, which was going on at the time.
Predicting the existence of eka-silicon, Mendeleev assumed that it would be found in minerals containing Ti, Zr, Nb, and Ta; he himself was going to analyse some rare minerals in search for the predicted element. Mendeleev, however, was not fated to do it and 15 years had to pass before eka-silicon was discovered.
In summer 1885, a new mineral was found in the Himmels–furst mine near Freiberg. It was named “argyrodite” since chemical analysis showed the presence of silver the Latin for which is argentum. The Freiberg Academy of Mining asked the chemist C. Winkler to determine the exact composition of the mineral. Analysis was comparatively easy and soon Winkler found the mineral to contain 74.72% silver, 17.43% Sulphur, 0.66% iron (II) oxide, 0.22% zinc oxide, and 0.31% mercury. But what surprised him was that the percentage of all the elements found in argyrodite added up to only 93.04 per cent instead of 100 per cent. No matter how many times Winkler repeated the analysis 6.96 per cent was missing.
Then Winkler made an assumption that the elusive amount had to be an unknown element. Inspired by the idea he began to study the mineral carefully and in February 1886 the principal events in the discovery of eka-silicon took place.
On February 6, Winkler reported to the German Chemical Society that he had succeeded in preparing some compounds of the new element and isolating it in a free state. The scientist’s report was published and sent to many scientific institutions all over the world. Here is the text received by the Russian Physico-Chemical Society: “The signatory has the honour to inform the Russian Physico-Chemical Society that he found in argyrodite a new non-metal element close in its properties to arsenic and antimony which he named “germanium”. Argyrodite is a new mineral found by Weisbach in Freiberg and consisting of silver, Sulphur, and germanium.”
Three points in this letter deserve attention: firstly, Winkler considered the new element to be a non-metal; secondly, he assumed its analogy with arsenic and antimony, and, thirdly, the element had already been named. Originally, Winkler wanted to name it “neptunium” but the name had already been given to another element–a false discovery–and the scientist proposed the name “germanium” after “Germany”. The name became widely accepted although not immediately.
Later it become clear that germanium is to a great extent amphoteric in nature and, hence, Winkler’s description of germanium as a non-metal cannot be considered completely erroneous. Much sharper debates revolved around the question the analogue of which element in the system germanium was. In his first report Winkler suggested arsenic and antimony but the German chemist Richter disagreed with Winkler saying that germanium, most likely, was identical to eka-silicon. Richter’s opinion seemed to affect the opinion of the discoverer of germanium and in his letter of February 26 to Mendeleev Winkler wrote: “At first I thought this element would fill the gap between antimony and bismuth in your remarkable and thoughtfully composed periodic system and that the element would coincide with your eka-antimony, but the facts indicate that here we are dealing with eka-silicon.”
Such as Winkler’s reply to Mendeleev’s letter of congratulation. It is interesting that the antimony-germanium analogy was considered erroneous by Mendeleev but he did not think of germanium as eka-silicon either. Probably, Mendeleev was surprised that the natural source of the new element proved to have nothing in common with that predicted by him earlier (titanium and zirconium ores). The discoverer of the periodic law proposed another hypothesis: germanium is an analogue of cadmium, namely eka–cadmium. It the nature of gallium and scandium was established beyond any doubt, as regads germanium, Mendeleev was less certain. This uncertainty, however, soon gave way to certainty and already on March 2 Mendeleev wired to Winkler conceding the identity of germanium and eka-silicon.
Soon an exhaustive article by Winkler entitled “Germanium–a new element” was published in the “Journal of Russian Physico-Chemical Society”. It was a new illustration of the brilliant similarity between the predicted properties of eka-silicon and real properties of germanium.
We have already briefly mentioned the discovery of scandium in the chapter devoted to REEs (see p. 130). Although many of scandium’s properties are similar to those of rare earths, D. I. Mendeleev predicted that the element would be a boron analogue in the third group of the periodic system. His prediction proved to be accurate enough. Scandium was discovered by the Swedish chemist L. Nilson; on March 12, 1879, his article “On Scandium, a New Rare Metal” was published and on March 24 it was discussed at a session of the Paris Academy of Sciences.
Nilson’s results, however, were in many respects erroneous. He considered scandium to be tetravalent and gave, therefore, the formula of its oxide as ScO2. He did not measure the atomic mass and gave only its probable range (160-180). And, finally, Nilson suggested that scandium should be placed in the periodic table between tin and thorium, which ran counter to Mendeleev’s prediction.
The discovery of scandium excited the scientific community and Nilson’s compatriot P. Cleve set out to study the newly discovered element. He studied it thoroughly for almost five months and came to the conclusion that many results obtained by Nilson were erroneous. Cleve reported to the Paris Academy of Science on August 18, and the academicians learnt much new about scandium. It turned out to be trivalent; its oxide’s formula was Sc2O; its properties differed somewhat from those determined by Nilson. According to Cleve (and this was especially important) scandium was the eka-boron predicted by Mendeleev; Cleve showed a table in the left-hand column of which eka-boron properties were given and in the right-hand one those of scandium the following day Cleve sent a letter to Mendeleev in which he wrote: “I have the honour to inform you that your element eka–boron, has been obtained”. It is scandium discovered by L. Nilson this spring.
And, finally, on September 10 Cleve published a long article about scandium from which it is clear that he had a much better understanding of the new element than Nilson. Therefore, those historians are who consider Cleve and Nilson as co–discoverer of scandium right.
For a long time Nilson was working under an illusion about some of scandium’s properties and refused to recognize its identity with eka-boron. Cleve’s investigations, however, impressed Nilson very much; in the long run he was forced to admit that he was wrong, thus doing justice to the prediction power of the periodic system.
All of Mendeleev’s predictions were confirmed in the long run. The last to be confirmed was the prediction of the density of metallic scandium; only in 1937 did the German chemistry W. Fischer succeed in preparing 98 per cent scandium. Its density was 3.0 g/cm3, that is exactly the figure predicted by Mendeleev.
The discovery of inert gases ranks among the four great scientific events of the end of the 19th century that led to revolutionary changes in natural sciences, the other three being the discovery of X-rays by Roentgen, discovery of radioactivity, and the discovery of electron.
This prominence given by scientists to inert gases has many reasons.The history of their discovery is colourful and exciting.
Helium, the mysterious solar element, was discovered on the earth and this fact alone illustrates how inventive and penetrating man’s mind became in his striving for deeper and better understanding of nature.
No less mysterious argon sowed confusion among scientists. Its chemical inertness made it impossible to be classified as a chemical element in the ordinary sense of the term since it revealed no chemical properties. There was nothing left for the researchers but to grow accustomed to the idea that there can be elements unable to enter into chemical reactions.
The idea proved extremely fruitful. The discovery of inert gases contributed to development of the zero valence concept. Moreover, forming an independent zero group they added harmony to the periodic system.
Almost twenty five years after their discovery the inert gases helped N. Bohr to develop his theory of the electron shells of atoms. This theory, in its turn, explained the chemical inactivity of the inert gases and their atomic structure became the basis of the concepts of ionic and covalent bonds. Thus,the discovery of inert gases contributed greatly to the development of theoretical chemistry.
In the early 60’s they surprised the scientific community once more. Scientists showed that Xenon (mainly) and krypton can form chemical compounds.Now more than 150 such compounds are known.
Inert gases are among the rarest stable elements on the earth.
Here are the data given by Ramsay: there is one part by volume of helium per 245 000 parts of atmospheric air, one of neon per 81 000 000, and one of argon per 106, one of krypton per 20 000 000 and one of xenon per 170 000 000. Since then these figures have remained almost unchanged. Ramsay said that xenon content in air is less than that of gold in sea water. This alone shows how excruciatingly difficult was the discovery of inert gases.
Krypton, Neon, and Xenon
In the history of inert gases, many problems stuck in starting. Out of many problems and their several reasons ; one of them was that scientists were dealing with very small amounts of Argon and Helium. To isolate them from air, one had to chemically remove oxygen, nitrogen, hydrogen, and carbon dioxide. All inert gases constitute a negligible part of the earth’s atmosphere.
Detection and isolation of so minor traces was difficult specially when Helium and Argon were known.
Another reason was chemical inactivity of Argon and Helium. Even the most active reagents (for instance, fluorine) were powerless to combine with these gases and isolate them . Chemists had no way of studying inert gases and only physical methods could bring some results. Therefore better physical methods were required and they were being developed during this discovery period.
Scientists developed experimental techniques for analyzing small amounts of gases, perfected spectroscopes and devices for determining gas densities.
Finally, an event took place that was of extreme importance for the history of inert gases. Two engineers, U. Hampson from England and G. Linde from Germany, invented and effective process for liquefaction of gases.Hampson built an apparatus that produced on liter of liquid air per hour.
In early 1898, M. Travers, Ramsay’s assistant, began to design a refrigerating apparatus for preparing large amounts of liquid argon. Since atmospheric gases liquefy at different temperatures, they can easily be separated from one another. The discoveries of argon and helium are remarkable also in that they set the chemists thinking not only about the nature of chemical inertness ( the phenomenon was understood only about a quarter of a century later) but about the periodic law and periodic system which were under a serious threat. Three most important characteristics of argon and helium (atomic masses, zero valence, monatomic molecule) put both gases outside the system. That is why Mendeleev was so readily attracted by the convenient thought about N3.
History has a striking power of prediction. Argon had not been properly discovered yet, when on May 24, 1894, Ramsay wrote a letter to Rayleigh in which he asked whether it had ever occurred to him that there was indeed a place in the periodic table for gaseous elements. For instance:
Li Be B C N O F X X X
Cl
Mn Fe Co Ni
Br
? Rd Ru Pd…
Ramsay assumed that the system’s small period could contain a triad to elements similar to those of iron and platinum metals in the great periods. The discoveries of argon and helium gave rise to an idea that these gases could occupy the places of two Xs in Ramsay’s graph. The atomic masses of these elements, however (4 and 40, respectively), proved to be too different for He and Ar to be placed in the same period. Gradually, the idea about new triads was relegated to the background and Ramsay proposed to place inert gases at the end of each period. In this case one could even expect the discovery of an element with the atomic mass 20, an intermediate between helium and argon. Ramsay’s report at the session of British Association in Toronto in August 1897, was devoted just to this element. The report was entitled “Undiscovered Gas”. Ramsay wanted to describe interesting properties of the gas but though it unwise not to mention its most remarkable property: the gas had not been discovered yet.
And here again we see the same certainty which permeated Ramsay’s letter to his wife on the eve of argon’s discovery. But not it was not audacity of a romantic but conviction multiplied by experience. The undiscovered gas turned out to be neon. Owing to a whim of fate (a frequent thing in science) the discovery was preceded by another event. The new gas could, obviously, be discovered by gradual evaporation of liquid air and by analysis of the resulting fractions, the ones lighter than argon being especially interesting. On May 24, 1898, Ramsay and Travers received a Dewar flask with liquid air. Unfortunately (or, rather, fortunately) the amount of air was too small to search for argon’s predecessor and the scientists decided to use the material for perfecting the procedure of liquid air fractionation. Having done so, Ramsay and Travers discovered by the end of the day that the fraction remained was the heaviest one. For a week the fraction remained neglected until on May 31 Ramsay decided to investigate it. The gas was scrubbed from possible impurities of nitrogen and oxygen and subjected to spectral analysis. Ramsay and Travers were dumbfounded when they saw a bright yellow line which could belong neither to helium nor sodium. Ramsay wrote down in this diary: “May 31. A new gas. Krypton.” Recall that this name was previously given to undiscovered helium. Now the name found its place in the history of inert gases. Krypton, however, was not the gas about which Ramsay made a report. Its density and atomic mass were higher than the predicted ones.
The discovery of neon promptly followed. Ramsay and Travers selected light fractions formed on the distillation of air and discovered a new inert gas in one of them. Ramsay later recollected that the name “neon” (from the Greek neos for “new”) had been proposed by Ramsay’s twelve-year-old son. In this case the experiment was performed by Travers alone since Ramsay was away. It was on the 7th of June. Then a whole week was required to confirm the result, obtain greater amounts of neon, and determine its density. Neon, as had been expected, turned out to be an intermediate between helium and argon although t had not yet been isolated as a pure gas. The problem of complete separation of neon and argon was solved later.
Still another inert gas was to be discovered by Ramsay and Travers. The scientists, however, did not feel as certain as in the case of neon. One day in July, 1898, the colleagues were busy with distilling liquid air and separating it into fractions. By midnight they collected more than 50 fractions discovering krypton in the last of them (No. 56). After that upon heating the apparatus one more fraction was collected (No. 57) consisting, mainly, of carbon dioxide traces. Ramsay and Travers argued about the expediency of studying it and at last decided to proceed with the of experiment. Next morning the scientists observed the spectrum of fraction No. 57, which turned out to be highly unusual. Ramsay and Travers concluded that it could be attributed to a new gas. pure xenon, however was prepared only in the middle of 1900. The name “xenon” originates from the Greek xenos, which means “stranger”
Argon
If you saw the statement “Inert gases were discovered by H. Cavendish in 1785” you would treat it as a joke. But no matter how paradoxical it seems, it is essentially true. Only the word “discovered” is misused here. One would be equally justified in declaring that hydrogen was discovered by R. Boyle in 1660 or by M.V Lomonosov in 1745. In his experiments Cavendish only observed “something” whose nature became clear on hundred years later. In one of his laboratory records Cavendish wrote that, passing an electric spark through a mixture of nitrogen with an excess of oxygen, he obtained a small amount of residue, no more than 1/125 the initial volume of the mixture. This mysterious gas bubble remained unchanged under the subsequent action of the electric discharge. It is clear now that it contained a mixture of inert gases, the fact which Cavendish could neither understand nor explain.
The famous English physicist’s experiment was described in 1849 by his biographer H. Wilson in the book Life of Herny Cavendish. In the early 80’s of the 19th century Ramsay studied the reaction of gaseous nitrogen with hydrogen and oxygen in the presence of a platinum catalyst. Nothing came out of these experiments and Ramsay did not even publish his results. As he recalled later, he had just read the book by Wilson and wrote “Pay attention” against the description of Cavendish’ experiment. He even asked his assistant C. Williams to repeat the experiment but we do not know the result of the attempt. Most likely, nothing came out of it. The episode, however, turned out to be unforgettable for Ramsay (his “hidden memory”, as he called it) and played a certain role in the prehistory of argon’s discovery. At first, the English physicist J. Rayleigh was the main character in it and the need for a further development of the atomic and molecular theory was its historic background. It was essential to specify the atomic masses of the elements for the development of the theory. Numerous experiments showed that in the majority of cases the atomic masses were not integers. Meanwhile, as early as 1815–1816 the English physician W. Prout advanced a hypothesis, a landmark in the history of natural sciences, that atoms of all chemical elements consist of hydrogen atoms; thus, atomic masses had to be integers. Therefore, either Prout was wrong, or the atomic masses were determined incorrectly.
To remove the discrepancy, new studies of the composition and nature of the gases were required. Rayleigh thought it necessary to determine, first of all, the densities of the main atmospheric gases, nitrogen and oxygen, since their atomic masses could then be calculated on the basis of the density values.
Rayleigh published a short article in the influential English journal Nature on September 29, 1892. It might seem that the article was about a mere trifle; the density of nitrogen separated from atmospheric air differed from that of nitrogen obtained by passing a mixture of air and ammonia over a red-hot copper wire. The difference was very small, only 0.001, but it could not be explained by an experimental error. Atmospheric nitrogen was heavier. Thus, a mystery appeared which was described as “an anomalously high density of atmospheric nitrogen”. Nitrogen obtained by any other chemical techniques was always lighter by the same value.
What was the cause of the discrepancy? Ramsay became interested in the problem. On April 19, 1894, he met with Rayleigh and discussed the situation. Each of them, however, remained firm in his previous conviction. Ramsay believed that atmospheric nitrogen contained an admixture of a heavier gas and Rayleigh, on the contrary, felt that an admixture of a lighter gas in “chemical” nitrogen was responsible for the discrepancy.
Rayleigh’s view seemed more attractive. The composition of atmosphere had been thoroughly studied for more than a hundred years and it was hardly possible that some components of the air could have remained undetected. It is just the time to remember Cavendish’s experiment and for Ramsay’s “hidden memory” to work. On April 29, Ramsay sent a letter to his wife in which he wrote that nitrogen, probably, contained some inert gas which had escaped their attention; Williams is combining nitrogen with magnesium and is trying to establish what remains after the reaction. “We can discover a new element.”
The latter breathes confidence: an unknown gas is a new element which, like nitrogen, is inactive, i.e., it hardly enters into chemical reactions. To separate the “stranger” from nitrogen, Ramsay tried to bond nitrogen chemically and used the reaction of nitrogen with red-hot magnesium shaving (3Mg+N2 = Mg3N2); this is the only example when chemistry played a role in the discovery of inert gases. Entering into polemics with himself Ramsay, however, assumed another possibility: the unknown gas is not a new element but an allotropic variety of nitrogen whose molecular consists of three atoms (N3) like oxygen (O2–molecular oxygen and O3–ozone). The absorption of nitrogen with magnesium must be accompanied with the decomposition of the N2 molecule into atoms; the single N atom could then be added to N2 forming N3. Such was Ramsay’s thinking and later the assumption about the existence of N3 became a trump card in the hands of argon’s opponents. Fruitless attempts to separate an ozone-like nitrogen continued for more than two months but by the 3rd of August Ramsay had 100 cm3 of a gas which was nitrogen with a density of 19.086.
The scientist wrote about his success to Crookes and Rayleigh. He send an ampoule with the gas Crookes for spectroscopic investigations; Rayleigh himself collected a small amount of the new gas. In the middle of August Ramsay and Rayleigh met at a scientific session and made a joint report. They described the spectrum of the gas and underlined its chemical inactivity. Many scientists listened to the report with interest but were surprised: how could it be that air contained a new component? The eminent physisist O. Lodge even asked: “Didn’t you, gentlemen, discover the name of the new gas as well?
The difficulty about the name was settled in early November when Ramsay suggested to Rayleigh to name it argon (from the Greek for “inactive”) taking into account its exceptional chemical inactivity and to assign the symbol A to it (which later became Ar.) On November 30, the president of the Royal Society Lord Kelvin (W. Thomson who in 1871 was the first to use the name “helium”) Publicly described the discovery of a new constituent of the atmosphere as the outstanding scientific event of the year. The nature of the constituent, however, was unclear. Was it a chemical element? Such authorities as D. I. Mendeleev and J. Dewar, the inventor of the flask for storage of liquid air, believed that argon was N3. The absolute chemical inactivity of argon was a new property previously unknown to chemists and, therefore, it was difficult to study the gas (in particular, to determine its atomic mass). In addition, it became clear that argon, unlike all known elemental gases, is monatomic, i.e. its molecule consists of one atom. At a session of the Russian Chemical Society on March 14, 1895, Mendeleev declared: argon’s atomic mass of 40 does not fit the periodic system, hence, argon is condensed nitrogen N3.
Much time had passed before the many problems presented by the discovery of argon were solved. A certain role was played here by the discovery of helium, which also turned out to be an inert and monatomic gas. The argon-helium pair allowed an assumption to be made that the existence of such gases is a regularity rather than a mere chance and one could expect the discovery of new representatives of this family. However, they were not discovered until three years passed. In the meantime scientists thoroughly studied the properties of helium and argon, made precise determination of their atomic masses, and put forward ideas about the location of both elements in the periodic table.
Helium’s unusual story attracted attention of many scientists and science historians, but the real sequence of events was distorted in numerous descriptions which overgrew with a lot of fictional details. Even a legend was invented beautiful and impressive–about the discovery of the sun element but it was far from the truth.
The French astronomer J. Janssen and the English astronomer N. Lockyer are considered to be the discoverers of helium. They studied the total solar eclipse of 1868 which was especially convenient to observe on the Indian ocean shores. In letters sent to the Paris Academy of Sciences and read out at one of its sessions they wrote that the spectra of the sun photographed during the eclipse contained a new yellow line D3 corresponding to an unknown element. To commemorate this remarkable event (the discovery of a new element existing on the sun but not on the earth) a special medal was minted.
Everything is wrong in this fascinating story except two dates. First of all, in August 1868, Lockyer was not on the Indian Ocean coast and did not observe the total solar eclipse. Janssen made his observations after the eclipse. They were of great importance for astronomy but not for the history of helium. The French astronomer was the first to observe solar prominences (gigantic ejections of solar matter) not during an eclipse and to describe their nature. Here is the text of the telegram sent by him to the Paris Academy of Science: “the eclipse and prominences were observed, the spectrum is remarkable and unexpected; Prominences are of a gaseous nature.”
Up to that time scientists had known nothing about the nature of prominences. Now it became clear they were clouds of gaseous matter and had a complex chemical composition. A detailed description of his observation was given by Janssen in a letter which reached Paris only 40 days later and was two weeks behind the letter of another French astronomer S. Raye. The latter also observed the prominences and made certain conclusions about them. And what was Lockyer doing at the time? Without leaving England, he observed the prominences with the help of a specially designed spectroscope and determined the positions of lines in their spectra. On October 23 he sent a letter to the Paris Academy of Sciences; by a surprising coincidence it was received on the same day as J. Janssen’s letter.
On October 26 the letters of Janssen and Lockyer were read to the session of the Academy but they did not contain a word about either the hypothetical sun element or the line which was later identified as the characteristic line of the helium spectrum. It was only pointed out in the letters that prominences had been observed when the sun was not eclipsed. And the medal was minted precisely to mark this event. Thus, no helium was discovered on August 18, 1868, either by Janssen or by Lockyer. Their observations provided an impetus for an intensive study of prominences by many astronomers. And only then was it noticed that the spectra of prominences contained a line which could be assigned to none of the elements known on the earth. Most clearly the line was observed by the Italian astronomer A. Secci who later designated it as D3. Secci’s name ought to be placed side by side with those of Janssen and Lockyer. His role in discovering helium was no less than that of his predecessors. Secci, however, assumed that the D3 line could belong to some known element, for instance, hydrogen, under high pressures and temperatures. If this assumption had not been confirmed, Secci would have agreed to consider D3 line as corresponding to some element unknown on Earth. N. Lockyer and E. Frankland tried to solve the problem posed by Secci but they did not notice any changes in the hydrogen spectrum. Therefore, in his article of April 3, 1871, Lockyer already used the expression “a new element X”. There are indications that the name “helium” (from the Greek helios for “solar”) was proposed by Frankland. The word “helium” was first uttered at a British Association Session by its president V. Thomsov (Lord Kelvin) on August 3, of the same year. Even if we regard the discovery of helium as “fait accompli”, then, it still remained unusual. It was the only element which could not be isolated in a material form. What is helium under ordinary conditions–gas, liquid, or solid? What are its properties? What is its atomic mass and where is its place in the natural series of elements?
None of these questions could be answered even approximately. Besides, Secci’s doubt was still not cleared. Thus, a period began in the history of helium when it was only a hypothetical element. There was no consensus on helium. Mendeleev firmly supported Secci’s point of view, feeling that the bright yellow line could belong to some other known element at high temperatures and pressures. W. Crookes, however, completely recognized helium’s independence and considered it to be a primary matter which gave rise to all other elements via successive transformations.
Sometimes it seemed that helium was not unique in its mysteriousness. Astronomers discovered new lines in the spectra of various cosmic objects: the sun, the stars, and nebulae. A number of hypothetical elements appeared, namely coronium, arconium, nebulium, protofluorine. Several years later they were all recognized to be nonexistent and only helium survived.
To receive recognition, helium had to show its “earth face” and its “earth” history began with a chance event.
On February 1, 1895, W. Ramsay received a short letter from K. Miers, a British museum employee. By that time Ramsay had already been acclaimed as the discoverer of argon and we may think Miers did not choose him by chance. Miers wrote about the experiments of the American researcher. W. Hildebrand, performed at the US Geological Institute as early as 1890. Upon heating of some thorium and uranium minerals (for instance, cleveite) a chemically inactive gas was liberated; its spectrum was similar to that of nitrogen and contained new lines.
Later Hilderbrand himself confessed to Ramsay that he had a temptation to attribute these lines to a new element. However, his colleagues were sceptical about the results and Hildebrand stopped his experiments. Miers, however, believed that in the light of numerous cases of nitrogen presence in natural uranates it was reasonable to stage another experiment.
Evidently, Ramsay believed that Hildebrand’s inactive gas could be argon; therefore, he agreed with Miers and on February 5, he acquired a small amount of cleveite. Ramsay himself, however, was busy with studying argon and attempting to prepare its compounds and, therefore, asked his pupil D. Matthews to carry out the experiment. Matthews treated the mineral with hot sulphuric acid and, like Hildebrand, observed the formation of bubbles of a gas resembling nitrogen.
When a sufficient amount of the gas was collected, Ramsay performed its spectral analysis (March 14). The picture was unexpected: the spectrum had a bright band whose lines were not found in the spectra of nitrogen and argon.
Although Ramsay had no sufficient facts to make definitive conclusions he assumed that cleveite contained, in addition to argon, another unknown gas. Ramsay spent a whole week to obtain this gas in as pure a form as possible. On March 22, he compared the spectra of argon and the unknown gas in the presence of B. Brauner. Ramsay provisionally named this gas “krypton” from the Greek for “secret”, “covered”. The name later passed to another inert gas. On March 23. The scientist wrote down in his diary that the bright yellow line of “krypton” did not belong to sodium and was not observed in the argon spectrum. (In the late sixties it was necessary to prove that the D3 line of solar helium was not the bright yellow line of sodium; history, as we see, repeated itself.)
Not quite sure of his result, Ramsay sent an ampoule with the gas to W. Crookes. A day later a telegram was received from Crookes which read: “krypton is helium, 587.49; come and see.” The figure 587.49 corresponded to the wavelength of the solar helium on a specially calibrated scale. Although these data facilitated the identification of helium on the earth, otherwise this discovery was independent. It became possible for the scientists to comprehensively study helium–a new chemical element which was no longer hypothetical. Helium’s complete chemical inactivity was not suspicious: similar inactivity of argon had already been known by that time (1894).
A brief communication about the discovery of helium on the earth was first published by Ramsay on March 29, 1895, in the “Chemical News” edited by Crookes. It is interesting that almost simultaneously terrestrial helium was discovered in cleveite by the Swedish scientist P. Cleve (in whose honour the mineral had been named) and by his assistant. A Lunglet. They, however, were a little too late with their experiments and could only express their disappointment, by no means claiming their priority. Terrestrial helium received full recognition and no attempts were made to refute Ramsay’s results. A little time passed and helium was discovered in other minerals and mineral spring waters. In 1898 helium was found in the earth atmosphere.
In the history of chemical elements the discovery of a new element often directly affected the discovery of another one. Thus, the discovery of thallium was a catalyst for the discovery of indium–the last of the classic group of four elements identified by spectral analysis.
The stage was set in the German town of Freiberg; and the main characters were F. Reich, professor of physics in the Mining Academy and his assistant Th. Richter. The time was the year of 1863. Interested in some properties if thallium, discovered two year earlier, F Reich decided to obtain a sufficient amount of the metal for his experiments. Searching for natural sources of thallium, he analysed samples of zinc ores mined at Himmelsfürst. In addition to zinc the ores were known to contain Sulphur, arsenic, lead, silicon, manganese, tin, and cadmium, in a word, quite a number of chemical elements. Reich believed that thallium could be added to the list. Although time-consuming chemical experiments did not produce the desired element, he obtained a straw-yellow precipitate of an unknown composition. It was told that when C. Winkler (subsequently the discoverer of germanium) entered Reich’s laboratory the latter showed him a test-tube with the precipitate and said that it contained sulphide of a new element.
It would have been surprising if F. Reich had not used spectroscopy to prove his assumption. Of course, Reich did use it but, unfortunately, he was colour-blind and, therefore, asked his assistant Richter to perform spectral analysis. Th. Richter succeeded in the very first attempt: in the spectrum of the sample he saw an extremely bright blue line which could not be confused with either cesium blue line or any other line. In a word, the observation was quite definite. Reich and Richter came to the conclusion that the ores of Himmelsfurst contained a new chemical element. They named it “indium” after “indigo”, a bright blue dye. There is an interesting fact that does credit to F. Reich. The first reports about the discovery of indium were signed by the two scientists. Reich, however, believed that this was unjust and that the honour of the discovery belonged solely to Richter.
Soon after the two scientists had proved the existence of natural indium with the help of spectroscopy, they obtained a small amount of it. Indium compounds turn the flame of a Bunsen burner blue-violet and so bright that presence of the new element could be established without a spectroscope. Subsequently Reich and Richter studied some properties of indium, with Winkler giving them considerable help.
When metallic indium, although contaminated, was prepared, Richter submitted the samples to the Paris Academy of Sciences in 1867 and estimated their value at 800 pounds sterling which was quite a lot of money at the time.
Chemical properties of indium were described soon after its discovery but its atomic mass was at first determined incorrectly (75.6). Mendeleev saw that this atomic mass would not correctly place indium in the periodic table and suggested to increase it by about 50 per cent. Mendeleev proved to be right and indium occupied its place in the third group of the periodic table.
Thallium became the third element whose presence in the earth minerals was established by spectroscopy. Some of its properties proved to be similar to those of alkali metals and, therefore, there were scientists who believed that thallium was not an independent chemical element but a mixture of alkali metals, namely unknown heavy analogues of rubidium and cesium. Time was required to dispel the doubts, while Bunsen and Kirchhoff continued to investigate the newly discovered elements their method of spectral analysis attracted attention of the English chemist and physicist W. Crookes. By that time, he had been known to the scientific community mainly as the editor and publisher of the Chemical News journal. There was nothing glamorous in the way Crookes started on his way to the discovery. Back in 1850 he received ten pounds of sludge remaining in lead chambers after production of sulphuric acid in Tilkerod plant (Germany). The scientist separated selenium from the sludge for the study of compounds called selenocyanides to which his first published paper was devoted. After the extraction of selenium and its purification a certain amount of the material remained ad there was every reason to suspect the presence of tellurium, a direct analogue of selenium in terms of chemical properties. However, with the methods he used to could not extract tellurium. The investigation was stopped and it was just a lucky chance that the scientist kept the residue after the processing of the sludge (and, perhaps, the belief that the residue contained tellurium).
The discovery of cesium and rubidium impressed W. Crookes very much. Being not only impressionable but practical as well, the scientist understood at once how very promising the spectral method was for analytical purposes. Having obtained a spectroscope, Crookes decided to test it immediately. The time came for the samples of the sulphuric acid sludge (or, to be more exact, its residue after removal of selenium) which had been kept for more than ten years. Crooks introduced the sample into the flame of a burner and was instantly disappointed: no hint of tellurium lines in the spectrum. The selenium lines appeared and the gradually faded. However instead of them a magnificent green line appeared which Crookes had never observed before. Of course, there was a temptation to assign the line to a new chemical element and the scientist did so naming it “thallium” from the Greek thallos, which means “a new green branch”.
The first publication about Crookes’ discovery appeared in Chemical News on March 30, 1861, under the title “On the Existence of a new Element Probably from the Sulphur Group”. Here the author was wrong since, as we know, thallium has nothing in common either with Sulphur or with its analogues. A year later Crookes recognized his mistake and published another paper titled “Thallium, a New Chemical Element” where no analogy with Sulphur was drawn. In this way was thallium discovered. The word “discovered” means here the establishing of the existence of thallium by the new method. After having observed the element’s spectrum Crookes neither separated the pure element nor prepared its compounds. This was done by the French chemist C. Lamy who is often credited with being an independent discoverer of thallium.
For the first time C. Lamy observed the green thallium line in a sample of selenium extracted from the sludge of sulphuric acid production (the raw material used by Crookes). This took place in March 1862, a year after Crookes’ observations, and already on June 23 Lamy submitted a sample of metallic thallium with a mass of about 14g to the Paris Academy of Science. Crookes also succeeded in preparing metallic thallium but in the form of powder. C. Lamy, however, declared that the thallium of Crookes was nothing other than the metal sulphide. Controversy went on. Crookes said that he had obtained the metal powder before May 1, 1862, but did not dare to fuse the powder into an ingot because of the product’s volatility. A special committee organized by the Paris Academy of Sciences, including such prominent scientists as A. Saint Claire Deville, T Pelouze, and J. Dumas, recognized the priority of G. Lamy.
The French chemist undoubtedly studied thallium in much greater detail than W. Crookes. He showed that the metal formed trivalent and monovalent compounds. Monovalent thallium has much in common with alkali metals; trivalent thallium resembles aluminium. J. Dumas named it “the paradoxical metal”. It was the similarity of thallium with sodium and potassium that gave rise to the idea that thallium was a mixture of unknown alkali metals with large atomic masses. It is regrettable that all the credit for the discovery of thallium is given to W. Crookes, while the French chemist’s significant achievements are often ignored.
In 1866 E. Nordenshöld, a well-known traveller, mineralogist and one of the explorers of Greenland, found a new mineral containing silver, copper, selenium, and thallium. He proposed to name it crookesite (in honour of W. Crookes). For a long time this mineral was believed to be the only one containing noticeable amounts of thallium.
The discovery of the second “spectral element” occurred in the studies of a rare mineral, lepidolite (called also lilalite because of its lilac colour). For the first time a detailed chemical analysis of lepidolite was performed by M. Klaproth at the end of the 18th century. But the experienced analyst did not discover alkalis in the mineral. Doubting his own results, Klaproth decided to repeat the analysis and this time (1797) he found the following components: 54.5% silicon dioxide, 38.25% aluminium oxide, 4% potassium oxide, and 0.75% manganese oxide. The missing 2.5 per cent Klaproth ascribed to the loss of water contained in the mineral. However, no matter what ingenious techniques the chemist tried, he could not determine the content of the two most important components: lithium (it had not been discovered yet by that time) and fluorine; thus, the nature of lepidolite remained obscure.
At the beginning of 1861 a sample of this mineral from Saxony fell into the hands of R. Bunsen and G. Kirchhoff, who separated alkaline components form it and precipitated potassium in the form of chloroplatinate. After a thorough washing the precipitate was subjected to spectral analysis. On February 23, 1861, the chemists reported the existence of a new alkali metal in lepidolite to the Berlin Academy of Sciences. The Scientists asserted that magnificent dark red colour of the line of the new metal gave them every reason to name the element “rubidium” and assign to it the symbol Rb from the Latin word rubidus, which meant a deep red colour. Then Bunsen and Kirchhoff discovered rubidium in the same mineral spring water in which cesium was found a year before. The rubidium content turned out to be only slightly higher than that of cesium. Metallic rubidium was prepared by R. Bunsen in 1863.

![\[6Ag+8HN{{O}_{3}}\to 6A{{g}^{+}}+2NO\uparrow +6NO_{3}^{-}+4{{H}_{2}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-ffc51e79178729609c3ac6f0e2923f3b_l3.png "Rendered by QuickLaTeX.com")
![\[2Ag+2{{H}_{2}}S{{O}_{4}}\to 2A{{g}^{+}}+SO_{4}^{2-}+S{{O}_{2}}\uparrow +2{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-bca35d1ca0d2ccf509f625a3e3e9a88f_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}^{+}}+C{{l}^{-}}\to AgCl\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-cd613f1889af0b147f46b97ecabbed76_l3.png "Rendered by QuickLaTeX.com")
![\[AgCl\downarrow +\,C{{l}^{-}}\rightleftharpoons {{[AgC{{l}_{2}}]}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-93f6b36f6bf3d6aed0d6817dce0eabbf_l3.png "Rendered by QuickLaTeX.com")
![\[AgCl\downarrow +2N{{H}_{3}}\to {{[Ag{{(N{{H}_{3}})}_{2}}]}^{+}}+C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-25ad92ef38a3405934297bda612dfe65_l3.png "Rendered by QuickLaTeX.com")
![\[AgCl\downarrow +2C{{N}^{-}}\to {{[Ag{{(CN)}_{2}}]}^{-}}+C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-6ce4238a620b96750953cde809f80d2a_l3.png "Rendered by QuickLaTeX.com")
![\[AgCl\downarrow +2{{S}_{2}}O_{3}^{2-}\to {{[Ag{{({{S}_{2}}{{O}_{3}})}_{2}}]}^{3-}}+C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-2963a109e75e987e55de6e25d6f26217_l3.png "Rendered by QuickLaTeX.com")
![\[2AgCl\downarrow \xrightarrow{(hv)}2Ag\downarrow +C{{l}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-a8967d1eeab83aaa2096d856cf6cd3a9_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}^{+}}+{{H}_{2}}S\to A{{g}_{2}}S\downarrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-4d5bbf46199f1dc7c3d941b7255f3649_l3.png "Rendered by QuickLaTeX.com")
![\[3A{{g}_{2}}S\downarrow +8HN{{O}_{3}}\to S\downarrow +2NO\uparrow +6A{{g}^{+}}+6NO_{3}^{-}+4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-29c24c10a45c8700f6235cee60526a87_l3.png "Rendered by QuickLaTeX.com")
![\[3A{{g}_{2}}S\downarrow +2HN{{O}_{3}}\to S\downarrow +2NO\uparrow +3A{{g}_{2}}O+{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-38037040e60456547b4d29ee21bb7510_l3.png "Rendered by QuickLaTeX.com")
![\[3A{{g}_{2}}S\downarrow +6HN{{O}_{3}}\to 6A{{g}^{+}}+6NO_{3}^{-}+3{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-30aebd3d6bbf492422370a8fbca7fbfb_l3.png "Rendered by QuickLaTeX.com")
![\[S\downarrow +2HN{{O}_{3}}\to SO_{4}^{2-}+2NO\uparrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-617688890df5f077d358eb619a5a7f9c_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}^{+}}+2N{{H}_{3}}+{{H}_{2}}O\to A{{g}_{2}}O\downarrow +2NH_{4}^{+}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-b528d24e09f3ebd8042a8cbd3e369523_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}O\downarrow +4N{{H}_{3}}+{{H}_{2}}O\to 2{{[Ag{{(N{{H}_{3}})}_{2}}]}^{+}}+2O{{H}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-9f1512dd0bfdf9708ab3ebfc232640aa_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}^{+}}+2O{{H}^{-}}\to A{{g}_{2}}O\downarrow +{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-e388651ef0e31a0512821c122056eb52_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}O\downarrow +{{H}_{2}}O\rightleftharpoons 2Ag{{(OH)}_{2}}\downarrow \,\rightleftharpoons 2A{{g}^{+}}+2O{{H}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-641fd0d3964912c51652aa1f28310cd0_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}O\downarrow +2{{H}^{+}}\to 2A{{g}^{+}}+{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-613454a27dc1ec2fcb395bfdbf33b7f2_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}^{+}}+{{I}^{-}}\to AgI\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-fd832c7900967e676733ce574bbc439b_l3.png "Rendered by QuickLaTeX.com")
![\[AgI+2C{{N}^{-}}\to {{[Ag{{(CN)}_{2}}]}^{-}}+{{I}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-fcc83e87b03592eba249da8de37b752b_l3.png "Rendered by QuickLaTeX.com")
![\[AgI+2{{S}_{2}}O_{3}^{2-}\to [Ag{{({{S}_{2}}{{O}_{3}}]}^{3-}}+{{I}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-63ce27e1f73019d7efbfd79d88634751_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}^{+}}+CrO_{4}^{2-}\to A{{g}_{2}}Cr{{O}_{4}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-f3501b2508735199c9203477464884fd_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}_{2}}Cr{{O}_{4}}\downarrow +2{{H}^{+}}\rightleftharpoons 4A{{g}^{+}}+C{{r}_{2}}O_{7}^{2-}+{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-4bab0203c91aa9ef1e67adc24579cd95_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}Cr{{O}_{4}}\downarrow +4N{{H}_{3}}\to 2{{[Ag{{(N{{H}_{3}})}_{2}}]}^{+}}+C{{r}_{2}}O_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-984fd908662409bb779e8a86fc1a7dfe_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}^{+}}+C{{N}^{-}}\to AgCN\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-883f33f245f8a0f7964c868527d37325_l3.png "Rendered by QuickLaTeX.com")
![\[AgCN\downarrow +C{{N}^{-}}\to {{[Ag{{(CN)}_{2}}]}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-48db9832b19ee9f0e68b1683280d9b99_l3.png "Rendered by QuickLaTeX.com")
![\[2A{{g}^{+}}+CO_{3}^{2-}\to A{{g}_{2}}C{{O}_{3}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-c1b78d65bda9d66d5c493954b7127c49_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}C{{O}_{3}}\downarrow \to A{{g}_{2}}O\downarrow +C{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-10922d0011deac01d4876d27c3ea3806_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}C{{O}_{3}}\downarrow +2{{H}^{+}}\to 2A{{g}^{+}}+C{{O}_{2}}\uparrow +{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-a32025b3bc15384fd6c6f77932cd6051_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{2}}C{{O}_{3}}\downarrow +4N{{H}_{3}}\to 2{{[Ag{{(N{{H}_{3}})}_{2}}]}^{2}}+CO_{3}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-bb5bd70c5380060f324affcea76381fc_l3.png "Rendered by QuickLaTeX.com")
![\[3A{{g}^{+}}+HPO_{4}^{2-}\to A{{g}_{3}}P{{O}_{4}}\downarrow +{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-3d1c7b68a692126e8ef252a3197eff5b_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{3}}P{{O}_{4}}\downarrow +3{{H}^{+}}\to 3A{{g}^{+}}+{{H}_{3}}P{{O}_{4}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-1c48f30d10cbc13ca4910634179e95d1_l3.png "Rendered by QuickLaTeX.com")
![\[A{{g}_{3}}P{{O}_{4}}\downarrow +6N{{H}_{3}}\to 3[Ag{{(N{{H}_{3}})}_{2}}+PO_{4}^{3-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-7de84c383e6acec27a8d8d0205e0de72_l3.png "Rendered by QuickLaTeX.com")
![\[4{{[Ag{{(N{{H}_{3}})}_{2}}]}^{+}}+{{H}_{2}}N-N{{H}_{2}}.{{H}_{2}}S{{O}_{4}}\to 4Ag\downarrow +{{N}_{2}}\uparrow +6NH_{4}^{+}+2N{{H}_{3}}+SO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-00f44badcaa383b16d5ac614837b83cd_l3.png "Rendered by QuickLaTeX.com")

![\[6Hg+8HN{{O}_{3}}\to 3Hg_{2}^{2+}+2NO\uparrow +6NO_{3}^{-}+4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-2a406a86905ebd050fe82189d2bbf868_l3.png "Rendered by QuickLaTeX.com")
![\[3Hg+8HN{{O}_{3}}\to 3H{{g}^{2+}}+2NO\uparrow +6NO_{3}^{-}+4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-0f3245649f75adfa9b9e5fda5a77defe_l3.png "Rendered by QuickLaTeX.com")
![\[2Hg+2{{H}_{2}}S{{O}_{4}}\to Hg_{2}^{2+}+SO_{4}^{2-}+S{{O}_{2}}\uparrow +2{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-85edbc7d5943a3841cef8bc6717fce3b_l3.png "Rendered by QuickLaTeX.com")
![\[Hg+2{{H}_{2}}S{{O}_{4}}\to H{{g}^{2+}}+SO_{4}^{2-}+S{{O}_{2}}\uparrow +2{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-57397010764439e35e3233386aebd5bd_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+2C{{l}^{-}}\to H{{g}_{2}}C{{l}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-2813b6f2e5f85f4d8cc06039d0b99059_l3.png "Rendered by QuickLaTeX.com")
![\[HgC{{l}_{2}}+2N{{H}_{3}}\to Hg\downarrow +Hg(N{{H}_{2}})Cl\downarrow +NH_{4}^{+}+C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-02a1a2eef0d7b6f315b4f10f3485f269_l3.png "Rendered by QuickLaTeX.com")
![\[3H{{g}_{2}}C{{l}_{2}}\downarrow +2HN{{O}_{3}}+6HCI\to 3HgC{{l}_{2}}+2NO\uparrow \text{ }+4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-71908886209f551410a2411fc652e352_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+{{H}_{2}}S\to \text{ }Hg\downarrow +HgS\downarrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-27fac1246f632b50ec4c57691e412df6_l3.png "Rendered by QuickLaTeX.com")
![\[HgS+{{S}^{2-}}\to {{[Hg{{S}_{2}}]}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-18749115f2284c168531e7d621ea07f0_l3.png "Rendered by QuickLaTeX.com")
![\[{{\left[ Hg{{S}_{2}} \right]}^{2-}}+2{{H}^{+}}\to HgS\downarrow +{{H}_{2}}S\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-5b569e67bc23d778cb4a6ba9ebfa8a77_l3.png "Rendered by QuickLaTeX.com")
![\[HgS\downarrow +Hg\downarrow +3S_{2}^{2-}\to 2{{[Hg{{S}_{2}}]}^{2-}}+S_{3}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-e9fcff8cd12e18be6f4cb8025926f05e_l3.png "Rendered by QuickLaTeX.com")
![\[Hg\downarrow +S_{2}^{2-}\to HgS\downarrow +{{S}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-f7f02853c35f8442ef851e9ae4ce4f4d_l3.png "Rendered by QuickLaTeX.com")
![\[HgS\downarrow +{{S}^{2-}}\to {{\left[ Hg{{S}_{2}} \right]}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-2122e487395393aa5259777783478d1b_l3.png "Rendered by QuickLaTeX.com")
![\[HgS+2S_{2}^{2-}\to HgS_{2}^{2-}+S_{3}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-c989628d1ae16f61a9f7f9629cf613eb_l3.png "Rendered by QuickLaTeX.com")
![\[12HCl+4HN{{O}_{3}},+3Hg\downarrow +3HgS\downarrow =6HgC{{l}_{2}}+3S\downarrow \text{ }+4NO\uparrow +8{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-5fac553afe7e05e9daa151da408d2053_l3.png "Rendered by QuickLaTeX.com")
![\[3HCI+HN{{O}_{3}}\to 3Cl+NO\uparrow +2{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-d8415db1b47296fb7432c306e1c83fcf_l3.png "Rendered by QuickLaTeX.com")
![\[HgS\downarrow +2CI\to HgC{{l}_{2}}+S\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-19f5b345e532eafcf50921516a5cee0a_l3.png "Rendered by QuickLaTeX.com")
![\[HgS\downarrow +2C1\to HgC{{l}_{2}}+S\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-05b4a59428355be06744f2c6fb337f99_l3.png "Rendered by QuickLaTeX.com")
![\[12HCl+4HN{{O}_{3}}+3Hg\downarrow +3HgS\downarrow =6HgC{{l}_{2}}+3S\downarrow +4NO\uparrow +8{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-7bbfe70a447b11824ccbfa937f60533e_l3.png "Rendered by QuickLaTeX.com")
![\[S\downarrow +6HCl+2HN{{O}_{3}}\to S_{4}^{2-}+6C{{l}^{-}}+8{{H}^{+}}+2NO\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-2e52276dde3a3009f743f1f0726d6b0b_l3.png "Rendered by QuickLaTeX.com")
![\[\begin{align} & \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,N{{H}_{2}} \\ & \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,/ \\ & 2Hg_{2}^{2+}+NO_{3}^{-}+4N{{H}_{3}}+{{H}_{2}}O\to HgO.Hg\,\,\,\,\,\,\,\downarrow +2Hg\downarrow +3NH_{4}^{+} \\ & \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\backslash \\ & \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,NO \\ \end{align}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-d64edb84c37d600cb06bb3b36d3cb695_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+2O{{H}^{-}}\to H{{g}_{2}}O\downarrow +{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-cf007f0a81e8e50f7dc67dff5fb27781_l3.png "Rendered by QuickLaTeX.com")
![\[H{{g}_{2}}O\downarrow \to HgO\downarrow +Hg\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-3d591ec36dd978abd26da4980dc654f0_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+CrO_{4}^{2-}\to H{{g}_{2}}Cr{{O}_{4}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-911d28e96d4e877260e8eb95ae75ecfd_l3.png "Rendered by QuickLaTeX.com")
![\[H{{g}_{2}}Cr{{O}_{4}}\downarrow +2O{{H}^{-}}\to H{{g}_{2}}O\downarrow +CrO_{4}^{2-}+{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-6a4c33a29a0b90ae7b1f8f979909fa42_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+2{{I}^{-}}\to H{{g}_{2}}{{I}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-0d4ffbad08f2e2855507286ca18082b0_l3.png "Rendered by QuickLaTeX.com")
![\[H{{g}_{2}}{{I}_{2}}\downarrow +2{{I}^{-}}\to {{[Hg{{I}_{4}}]}^{2-}}+Hg\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-5c7a5eaefb396ac8392e3d7fd5b3a0bc_l3.png "Rendered by QuickLaTeX.com")
![\[H{{g}_{2}}{{I}_{2}}\downarrow \to Hg{{I}_{2}}\downarrow +Hg\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-0fa61634ee3ed2d83f3784681321b88d_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+CO_{3}^{2-}\to H{{g}_{2}}C{{O}_{3}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-71ab18bf9d27b4da8712664f04c2ca16_l3.png "Rendered by QuickLaTeX.com")
![\[H{{g}_{2}}C{{O}_{3}}\downarrow \to HgO\downarrow +Hg\downarrow +C{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-097261c4156fd62322f895a09841d4e8_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+HPO_{4}^{2-}\to H{{g}_{2}}HP{{O}_{4}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-113d02b1e10b0de7d14cf42d71905897_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+2C{{N}^{-}}\to Hg\downarrow +Hg{{(CN)}_{2}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-212d091c8e981537c07cf1d41f2ee648_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+S{{n}^{2+}}\to 2Hg\downarrow +S{{n}^{4+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-094ff1c478e5aeeb451f75eb562662a9_l3.png "Rendered by QuickLaTeX.com")
![\[Hg_{2}^{2+}+NO_{2}^{-}+{{H}_{2}}O\to 2Hg\downarrow +NO_{3}^{-}+2{{H}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-1b391191ceba70658ade3195fcc253dd_l3.png "Rendered by QuickLaTeX.com")
![\[Cu+Hg_{2}^{2+}\to C{{u}^{2+}}+2Hg\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-98e418cec0856eb674cb6148ab0a6be5_l3.png "Rendered by QuickLaTeX.com")
![\[3Hg_{2}^{2+}+2Al\to 2A{{l}^{3+}}+6Hg\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-575a14736ff8d7ae0f6771cb345021c6_l3.png "Rendered by QuickLaTeX.com")

![\[3Pb+\text{ }8HN{{O}_{3}}\text{ }\to \text{ }3P{{b}^{2+}}+\text{ }6NO_{3}^{-}\text{ }+2NO\uparrow +4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-774f29e693f92e07f0e5acff113b6d15_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+2C{{l}^{-}}\Leftrightarrow PbC{{l}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-369319e1bf7a15a7c7c3859b3a43ddb2_l3.png "Rendered by QuickLaTeX.com")
![\[PbC{{l}_{2}}\downarrow +2C{{l}^{-}}\to {{[PbC{{l}_{4}}]}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-c593a9dfee151fa50e16c12f547f3492_l3.png "Rendered by QuickLaTeX.com")
![\[PbC{{l}_{2}}\downarrow +2N{{H}_{3}}+2{{H}_{2}}O\to \text{ }Pb{{\left( OH \right)}_{2}}\downarrow +2NH_{4}^{+}\text{+}2C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-ac70cc97de78e1f6cb786827e3cbba50_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+{{H}_{2}}S\to PbS\downarrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-8ce539e85d66a1c70a79074c9b82b8ac_l3.png "Rendered by QuickLaTeX.com")
![\[PbC{{l}_{2}}\downarrow {{H}_{2}}S\to PbS\downarrow +2{{H}^{+}}+2C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-62e89e8bd0011b9789f08f4547bca4a9_l3.png "Rendered by QuickLaTeX.com")
![\[2P{{b}^{2+}}+{{H}_{2}}S+2C{{l}^{-}}\to P{{b}_{2}}SC{{l}_{2}}\downarrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-9315a8d6aa16482cd1e266d5fd3f3312_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}_{2}}SC{{l}_{2}}\downarrow \to PbS\downarrow +PbC{{l}_{2}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-fc3d30f06bca322b51d44cbfba44a0e7_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}_{2}}SC{{l}_{2}}\downarrow +{{H}_{2}}S\to 2PbS\downarrow +2C{{l}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-4fcbc48b723de0bd428a90cd08253b77_l3.png "Rendered by QuickLaTeX.com")
![\[3PbS\downarrow +8HN{{O}_{3}}\to 3P{{b}^{2+}}+6NO_{3}^{-}+3S\downarrow +2NO\uparrow +4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-e9dc6d4490868674cc8d70f067a888d9_l3.png "Rendered by QuickLaTeX.com")
![\[S\downarrow +2HN{{O}_{3}}+SO_{4}^{2-}+2{{H}^{+}}+2NO\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-da9a23cdc866e032f4a4b8b26cc8e581_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+SO_{4}^{2-}+2{{H}^{+}}+2NO\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-c51e0ac815c963de58b4004c6f49ef80_l3.png "Rendered by QuickLaTeX.com")
![\[PbS\downarrow +4{{H}_{2}}{{O}_{2}}\to PbS{{O}_{4}}\downarrow +4{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-9fac1560cead1bb45b47d6fb8c863f23_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+2N{{H}_{3}}+2{{H}_{2}}O\to Pb{{(OH)}_{2}}\downarrow +2NH_{4}^{+}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-b575a3f2e02201bf876835bb1b07e7a7_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+2O{{H}^{-}}\to Pb{{(OH)}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-349ffb7b7d640146d432f295a43ca7a0_l3.png "Rendered by QuickLaTeX.com")
![\[Pb{{(OH)}_{2}}\downarrow +2O{{H}^{-}}\to {{[Pb{{(OH)}_{4}}]}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-724799f6930565e29bd77e13e6772521_l3.png "Rendered by QuickLaTeX.com")
![\[{{[Pb{{(OH)}_{4}}]}^{2-}}+{{H}_{2}}{{O}_{2}}\to Pb{{O}_{2}}\downarrow +2{{H}_{2}}O+2O{{H}^{-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-b5c0bffac258bd6e68e0288687994a27_l3.png "Rendered by QuickLaTeX.com")
![\[{{[Pb{{(OH)}_{4}}]}^{2-}}+{{S}_{2}}O_{8}^{2-}\to Pb{{O}_{2}}\downarrow +2{{H}_{2}}O+2SO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-52fb1ff553284c86d06b726f16e5d29b_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+SO_{4}^{2-}\to PbS{{O}_{4}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-f7e2d1406ddea5ed06144c232435b368_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{4}}\downarrow +{{H}_{2}}S{{O}_{4}}\to P{{b}^{2+}}+2HSO_{4}^{-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-e85426f12b047270544dc7117d13e3eb_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{4}}\downarrow +\,4C{{H}_{3}}CO{{O}^{-}}\to {{[Pb{{(C{{H}_{3}}COO)}_{4}}]}^{2-}}+\,\,SO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-631140c3d73cebffaaa0768dd71dd6ff_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{4}}\downarrow +\,2{{C}_{4}}{{H}_{4}}O_{6}^{2-}\to [Pb{{({{C}_{4}}{{H}_{4}}{{O}_{6}}]}^{2-}}+\,\,SO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-aaf581647cbb54cf22ee9851d00038cb_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{4}}\downarrow +CO_{3}^{2-}\to PbC{{O}_{3}}\downarrow +SO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-dc3a4d511601ff5974e4f41677404020_l3.png "Rendered by QuickLaTeX.com")
![\[PbC{{O}_{3}}\downarrow +2{{H}^{+}}\to P{{b}^{2+}}+{{H}_{2}}O+C{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-0f186387adeb9ad4f79eceaf86a645be_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+CrCO_{4}^{2-}\to PbCr{{O}_{4}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-12d25c97925c8089b722cfcfac43bf6c_l3.png "Rendered by QuickLaTeX.com")
![\[2PbCr{{O}_{4}}\downarrow +2{{H}^{+}}\rightleftarrows 2P{{b}^{2+}}+C{{r}_{2}}O_{7}^{2-}+2{{H}_{2}}O\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-9071d8dfa23b27daed9aadaabefac590_l3.png "Rendered by QuickLaTeX.com")
![\[PbCr{{O}_{4}}\downarrow +4O{{H}^{-}}\rightleftarrows {{[Pb{{(OH)}_{4}}]}^{2+}}+CrO_{4}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-3b919f50bc13983557c1606172d0367f_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+2{{I}^{-}}\to Pb{{I}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-69ca27fb5f83a307d53613422252e899_l3.png "Rendered by QuickLaTeX.com")
![\[Pb{{l}_{2}}\downarrow +2{{I}^{-}}\rightleftarrows {{[Pb{{I}_{4}}]}^{2-}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-f1a130aa86944d00f00e8f53a59f4e95_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+SO_{3}^{2-}\to PbS{{O}_{3}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-c14ef40c3e00299545d7db39b028171e_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{3}}\downarrow +2{{H}^{+}}\to P{{b}^{2+}}+{{H}_{2}}O+S{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-24c8a4e43f4835a0d93095d810e90afe_l3.png "Rendered by QuickLaTeX.com")
![\[PbS{{O}_{3}}\downarrow +4O{{H}^{-}}\to {{[Pb{{(OH)}_{4}}]}^{2-}}+SO_{3}^{2-}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-a728d2f68fbe684b8694492934b9eead_l3.png "Rendered by QuickLaTeX.com")
![\[2P{{b}^{2+}}+2CO_{3}^{2-}+{{H}_{2}}O\to Pb{{(OH)}_{2}}\downarrow +PbC{{O}_{3}}+C{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-9e5f3855331d1d889a8d483a2b95a3af_l3.png "Rendered by QuickLaTeX.com")
![\[Pb{{(OH)}_{2}}\downarrow +PbC{{O}_{3}}\downarrow +4{{H}^{+}}\to 2P{{b}^{2+}}+3{{H}_{2}}O+C{{O}_{2}}\uparrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-3098b6c8741cf3b550fd0580c5cf3219_l3.png "Rendered by QuickLaTeX.com")
![\[3P{{b}^{2+}}+2HPO_{4}^{2-}\rightleftarrows \,\,P{{b}_{3}}{{(P{{O}_{4}})}_{2}}\downarrow +2{{H}^{+}}\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-b246205d2506362cd9e28bf93369d4f7_l3.png "Rendered by QuickLaTeX.com")
![\[P{{b}^{2+}}+2C{{N}^{-}}\to Pb{{(CN)}_{2}}\downarrow \]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-b3985cfa4f0cffdb707a96d4827e7124_l3.png "Rendered by QuickLaTeX.com")



![\[^{239}Pu+n{{\to }^{240}}Pu+n{{\to }^{241}}Pu{{\xrightarrow{{{\beta }^{-}}}}^{241}}95+n{{\to }^{242}}95{{\xrightarrow{{{\beta }^{-}}}}^{242}}96\]](https://thechemistryguru.com/wp-content/ql-cache/quicklatex.com-6e156291894f8dafe81771fe1fe7eaa9_l3.png "Rendered by QuickLaTeX.com")